Log on to https://ondemand.ccr.buffalo.edu
-
Set up RStudio for today
source('/projects/rpci/rplect/Rpg520/Rsetdirs.R') -
Let RStudio know about your new working directory
Introduction
We use two case studies today. The studies are used to illustrate the data manipulation and statistical analysis tasks that one might expect in day-to-day wet- or dry-lab work in computational oncology.
US CDC Behavioral Risk Factor Surveillance System survey data
We use data from the US Center for Disease Control’s Behavioral Risk Factor Surveillance System (BRFSS) annual survey. Check out the web page for a little more information. We are using a small subset of this data, including a random sample of 20000 observations from each of 1990 and 2010.
In this first case study, We will use functions from the dplyr package to
manage our data. dplyr is is a key
package in a particular approach to data management in R called
the ‘tidyverse’. Tidy analysis uses a standard data structure (a
tibble) and relatively small number functions to provide an
effective way to accomplish many tasks.
Start by loading the dplyr package.
Functions for ‘tidy’ data management include:
-
filter()– filter data to contain specific rows. -
select()– select a subset of columns. -
mutate()– change or add columns.
Example functions to summarize data:
-
count()– simple count of entries in each column. -
summarize()– e.g.,mean()orvar()iance of one or more columns. -
group_by()– summarize data by group.
tidyr and other packages complement basic functionality
-
tidyr::pivot_wider()– change a ‘long’ table to a ‘wide’ table. See example below.
We will use the readr package to read data from disk into R.
tidyr and readr are used less commonly, so we do not load them into our R session yet.
For visualization, we use ggplot2; details are provided below.
Finally, for statistical functions we use the ‘stats’ package distributed with R.
Data input
Use file.choose() to find the ‘BRFSS-subset.csv’ file on
the CCR file system.
brfss_file_path <- file.choose() # look for 'Rpg520/week-04/extdata/BRFSS-subset.csv'Take a peak at the data using the readLines() function
to read the first 3 lines of data, and cat("\n") to print
the result to the terminal
readLines(brfss_file_path, 3) |>
cat(sep = "\n")
## "Age","Weight","Sex","Height","Year"
## 31,48.9879760010809,"Female",157.48,"1990"
## 57,81.6466266684681,"Female",157.48,"1990"The file is a ‘comma-separated value’ file. These files can be created by, for instance, using Excel’s ‘Export’ menu. The first line consists of column headers, separated by a comma. Subsequent lines represent rows of data, again with a comma separating columns.
Read the entire dataset into R with the command
readr::read_csv(). Assign the data to a variable
brfss.
brfss <- readr::read_csv(brfss_file_path)
## Rows: 20000 Columns: 5
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: ","
## chr (1): Sex
## dbl (4): Age, Weight, Height, Year
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
brfss
## # A tibble: 20,000 × 5
## Age Weight Sex Height Year
## <dbl> <dbl> <chr> <dbl> <dbl>
## 1 31 49.0 Female 157. 1990
## 2 57 81.6 Female 157. 1990
## 3 43 80.3 Male 178. 1990
## 4 72 70.3 Male 170. 1990
## 5 31 49.9 Female 155. 1990
## 6 58 54.4 Female 155. 1990
## 7 45 69.9 Male 173. 1990
## 8 37 68.0 Male 180. 1990
## 9 33 65.8 Female 170. 1990
## 10 75 70.8 Female 152. 1990
## # ℹ 19,990 more rowsInitial cleaning
The data are pretty simple, but two small changes will make it more
useful. Both ‘Sex’ and ‘Year’ are really factor values
(each can only take on specific levels, ‘Female’ and ‘Male’ for ‘Sex’,
and ‘1990’ and ‘2010’ for ‘Year’).
Mutate
A factor can be created from an ordinary vector x with
code like the following:
x <- c("Male", "Female", "Female", NA)
factor(x, levels = c("Female", "Male"))
## [1] Male Female Female <NA>
## Levels: Female Male
y <- c(2010, 1991, 1990, NA)
factor(y, levels = c("1990", "2010"))
## [1] 2010 <NA> 1990 <NA>
## Levels: 1990 2010Use the dplyr mutate() function to change ‘Sex’ and
‘Year’ to factors.
brfss |>
mutate(
Sex = factor(Sex, levels = c("Female", "Male")),
Year = factor(Year, levels = c("1990", "2010"))
)
## # A tibble: 20,000 × 5
## Age Weight Sex Height Year
## <dbl> <dbl> <fct> <dbl> <fct>
## 1 31 49.0 Female 157. 1990
## 2 57 81.6 Female 157. 1990
## 3 43 80.3 Male 178. 1990
## 4 72 70.3 Male 170. 1990
## 5 31 49.9 Female 155. 1990
## 6 58 54.4 Female 155. 1990
## 7 45 69.9 Male 173. 1990
## 8 37 68.0 Male 180. 1990
## 9 33 65.8 Female 170. 1990
## 10 75 70.8 Female 152. 1990
## # ℹ 19,990 more rowsThat looks like it’s working, save the updated data set
Data exploration
A good place to start with an analysis is basic data exploration. Perhaps the most straight-forward thing to do is count the number of observations in each year.
Count
brfss |>
count(Year)
## # A tibble: 2 × 2
## Year n
## <fct> <int>
## 1 1990 10000
## 2 2010 10000The data has been chosen so that each year has the same number of individuals. What about the number of females and males in each year? This is determined by responses to the survey, reflecting the relative number of males and females in the population, or at least responding to the survey.
brfss |>
count(Sex)
## # A tibble: 2 × 2
## Sex n
## <fct> <int>
## 1 Female 12039
## 2 Male 7961What about the number of each sex in each year? Use
count() with two (or more) column names
brfss |>
count(Sex, Year)
## # A tibble: 4 × 3
## Sex Year n
## <fct> <fct> <int>
## 1 Female 1990 5718
## 2 Female 2010 6321
## 3 Male 1990 4282
## 4 Male 2010 3679It seems like there are more Female respondents in 2010 than in 1990.
Use tidyr‘s
function pivot_wider() (remember to look at the help page
?pivot_wider for details on how this function works) to
pivot the ’Sex’ column entries to column names.
brfss |>
count(Sex, Year) |>
tidyr::pivot_wider(names_from = "Sex", values_from = "n")
## # A tibble: 2 × 3
## Year Female Male
## <fct> <int> <int>
## 1 1990 5718 4282
## 2 2010 6321 3679Summarize
We used tidyr::pivot_wider() instead of just
pivot_wider(). This notation means ’use the tidyr package, and a
function in that package called pivot_wider(). This
notation to avoid conflicts if two packages have a function named
pivot_wider(); it also has additional benefits related to
managing R’s .GlobalEnv, but that is a more
advanced topic.
Use summarize() for summaries more complicated than
simple counts
brfss |>
summarize(
avg_age = mean(Age, na.rm = TRUE),
ave_wt = mean(Weight, na.rm = TRUE),
ave_ht = mean(Height, na.rm = TRUE)
)
## # A tibble: 1 × 3
## avg_age ave_wt ave_ht
## <dbl> <dbl> <dbl>
## 1 51.0 75.4 169.Nothing too exciting here, except to perhaps note the use of metric system Weight (kg) and Height (cm).
The function mean() calculates the average of the
corresponding column. na.rm = TRUE tells the function to
remove NA (missing) values before calculating the mean.
This is kind of interesting. Suppose one had a vector and calculated the
mean
No surprises. What if one of the values in x were
missing? The logic used by R is that the missing value could be
any number, so the mean could be anything – if there’s
an unknown value, then the mean must also be unknown!
Often we would like to calculate the mean after removing the unknown
values, and this is what the na.rm = TRUE argument does
mean(x, na.rm = TRUE)
## [1] 2Group
Back to our data exploration, we might expect the average age,
weight, and height to be different between Female and Male respondents,
and perhaps also between years. Use group_by() to calculate
summaries by Sex and Year
brfss |>
group_by(Sex, Year) |>
summarize(
avg_age = mean(Age, na.rm = TRUE),
ave_wt = mean(Weight, na.rm = TRUE),
ave_ht = mean(Height, na.rm = TRUE)
)
## `summarise()` has grouped output by 'Sex'. You can override using the `.groups`
## argument.
## # A tibble: 4 × 5
## # Groups: Sex [2]
## Sex Year avg_age ave_wt ave_ht
## <fct> <fct> <dbl> <dbl> <dbl>
## 1 Female 1990 46.2 64.8 163.
## 2 Female 2010 57.1 73.0 163.
## 3 Male 1990 43.9 81.2 178.
## 4 Male 2010 56.2 88.8 178.This shows some interesting aspects of the data. Males are on average 15cm taller than females; there is no difference in average height between years.
The average age of both Female and Male respondents is greater in 2010 than in 1990. This likely reflects changing demographics, as the ‘baby boom’ cohort ages. Note that the average Female age changes by about 10.9 years, whereas the average Male age changes by about 12.3 years; perhaps this reflects different life expectancy of males and females.
Also interesting is that the average weight changes, by about 8.2 kg (18 lbs) for Female respondents, 7.6 kg (16.7 lbs) for Males. This could be because people in general have become heavier, or that older people are heavier than younger people (or for other reasons not captured in the data).
Visual exploration
We will use the ggplot2 package to visually explore our data. We use several functions from this package, so load it into our R session.
ggplot2 constructs plots by adding layers. Layers have different roles. The plot starts with the specification of the data set to be plotted…
Aesthetics & geometries: box plot, histogram, density plot
ggplot(brfss_2010)…and then adds the ‘aesthetics’ (columns to be used for X and Y axes, how to color points, etc)
…and finally one or more ‘geometries’ (‘geom’) that describe the
geometric relationship between the x and y aesthetics. One geom is
geom_boxplot(), which draws a box-and-whiskers plot.
ggplot(brfss_2010) +
aes(x = Sex, y = Weight) +
geom_boxplot()
## Warning: Removed 395 rows containing non-finite outside the scale range
## (`stat_boxplot()`).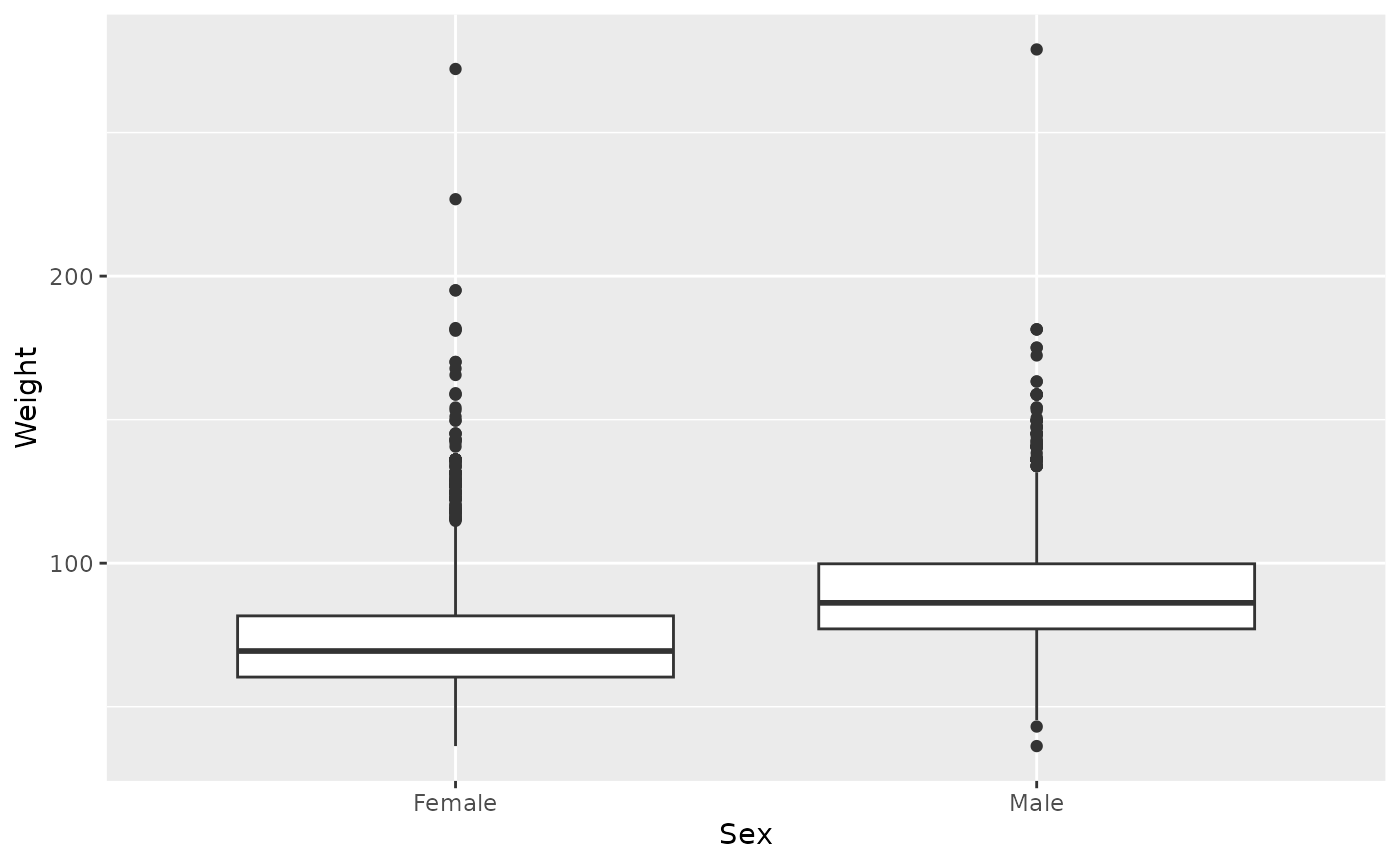
The bar in the figure is the median weight, the box represents upper and lower quartile of the data, the whiskers extend to 1.5 times the inter-quartile range. Points outside the whiskers represent potential outliers. The figure shows the difference in Female and Male weights, as well as a skew in the weight distribution of both Female and Male respondents.
Another way to view this sort of data is as a histogram…
brfss_2010 |> filter(Sex == "Male") |>
ggplot() +
aes(x = Weight) +
geom_histogram(col = "white")
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
## Warning: Removed 49 rows containing non-finite outside the scale range
## (`stat_bin()`).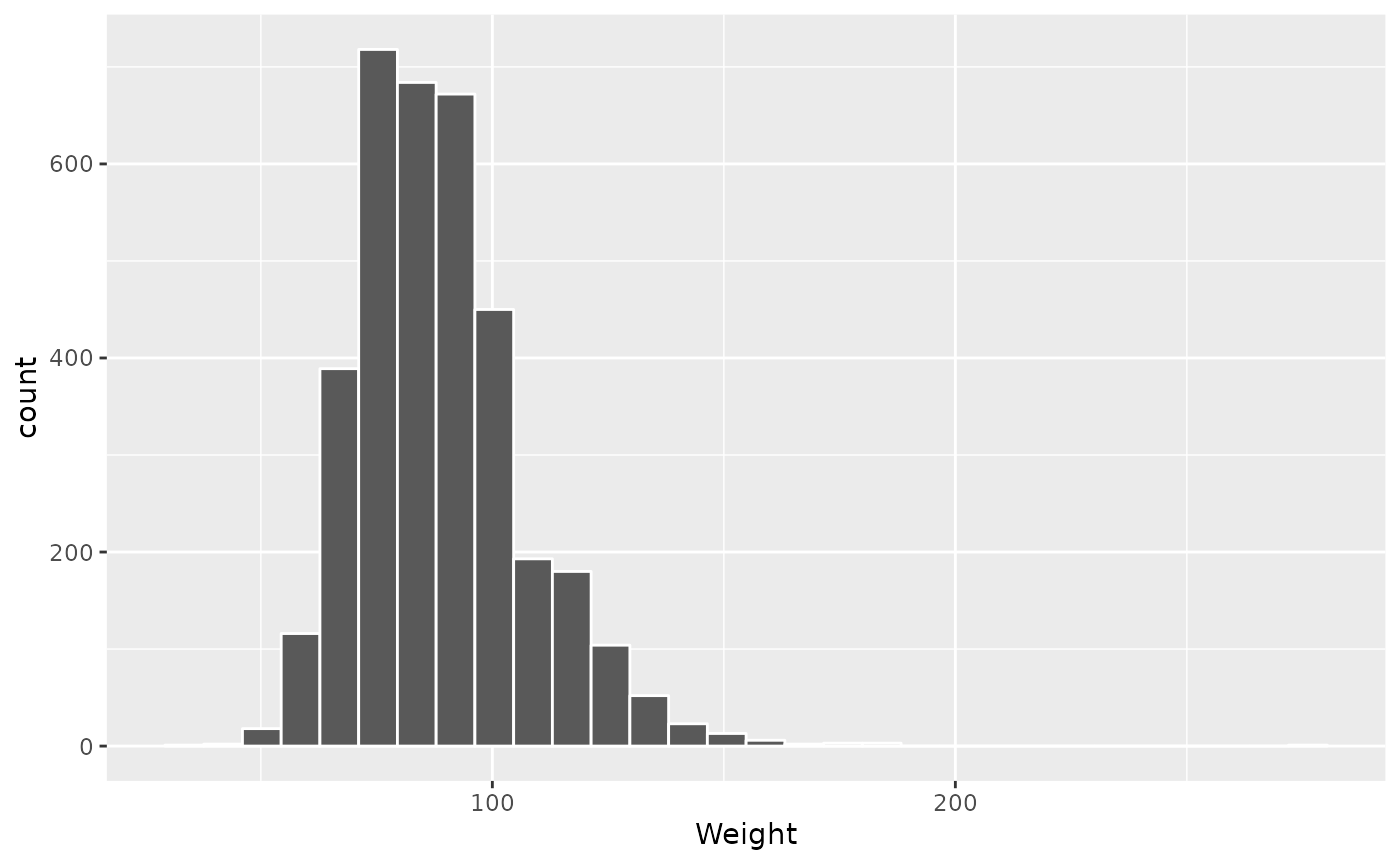
… or when comparing distributions as a density plot
ggplot(brfss_2010) +
aes(x = Weight, color = Sex) +
geom_density()
## Warning: Removed 395 rows containing non-finite outside the scale range
## (`stat_density()`).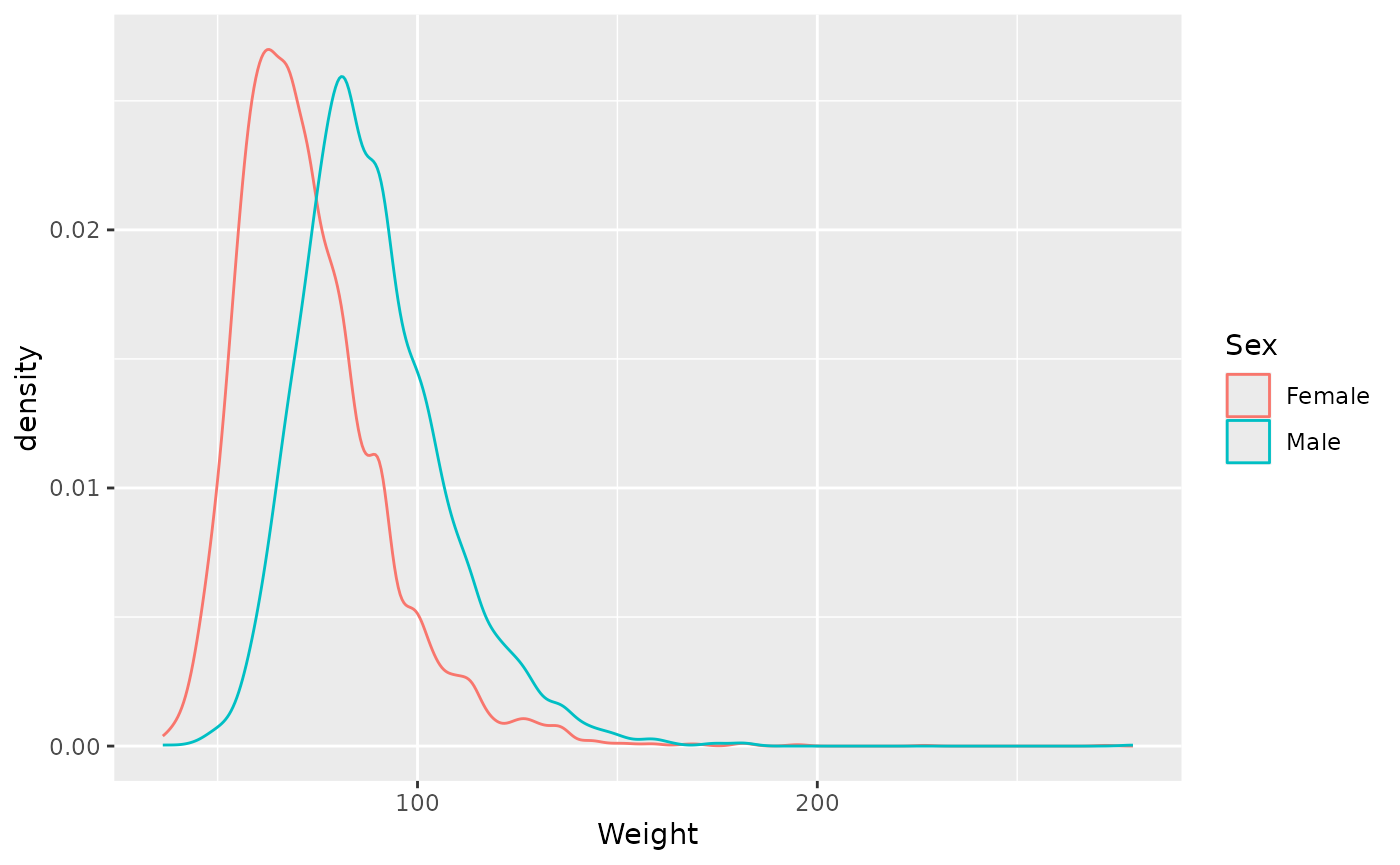
For subsequent work, note that log-transformed Weight reduces the most extreme values so the distribution is more Normal.
ggplot(brfss_2010) +
aes(x = log10(Weight), color = Sex) +
geom_density()
## Warning: Removed 395 rows containing non-finite outside the scale range
## (`stat_density()`).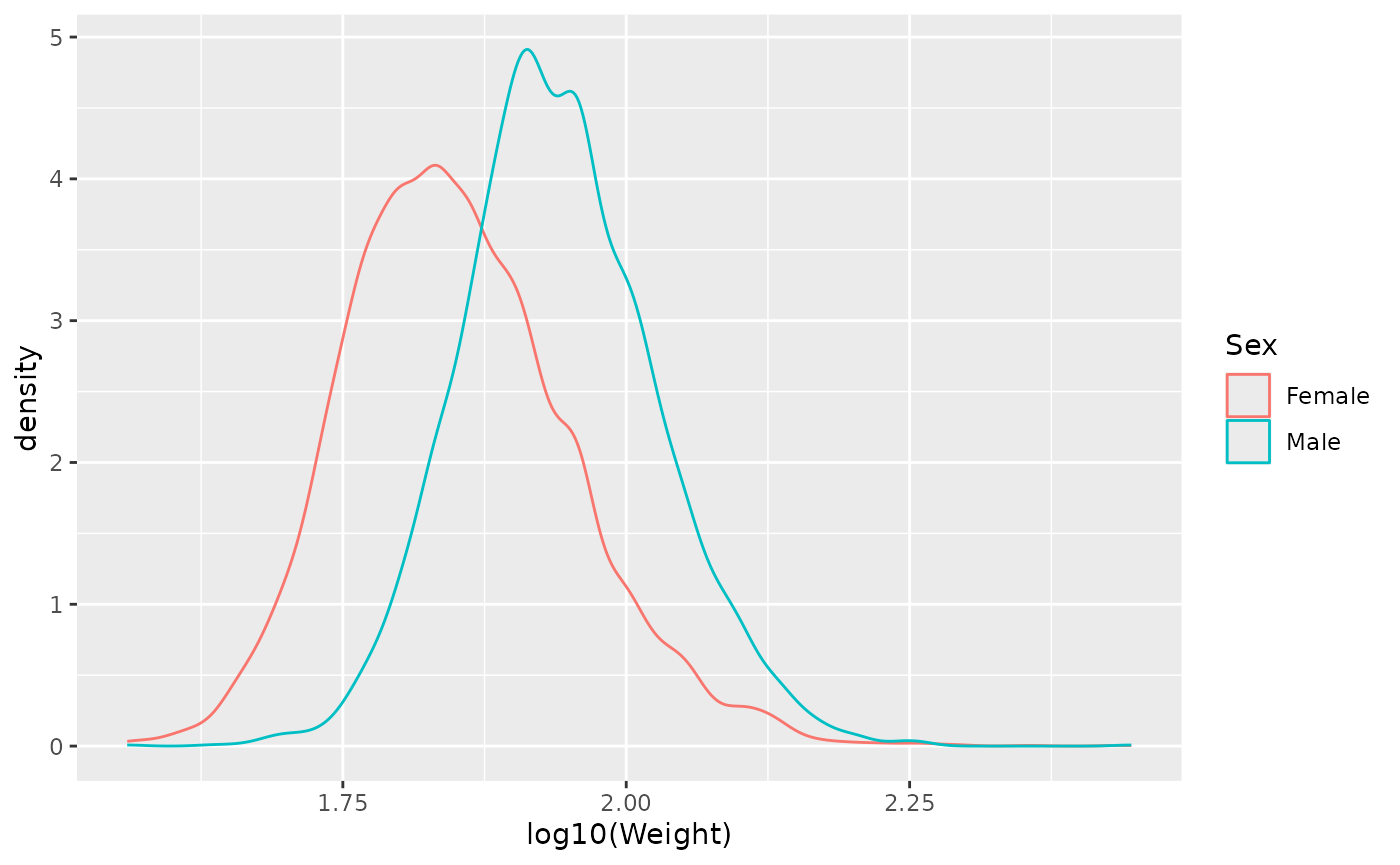
Scatter plots
Presumably taller people are heavier than shorter people. Replace
‘Sex’ with ‘Height’ in aes(), and replace
geom_boxplot() with geom_point() to generate a
scatter plot showing this relationship
ggplot(brfss_2010) +
aes(x = Height, y = Weight) +
geom_point()
## Warning: Removed 450 rows containing missing values or values outside the scale range
## (`geom_point()`).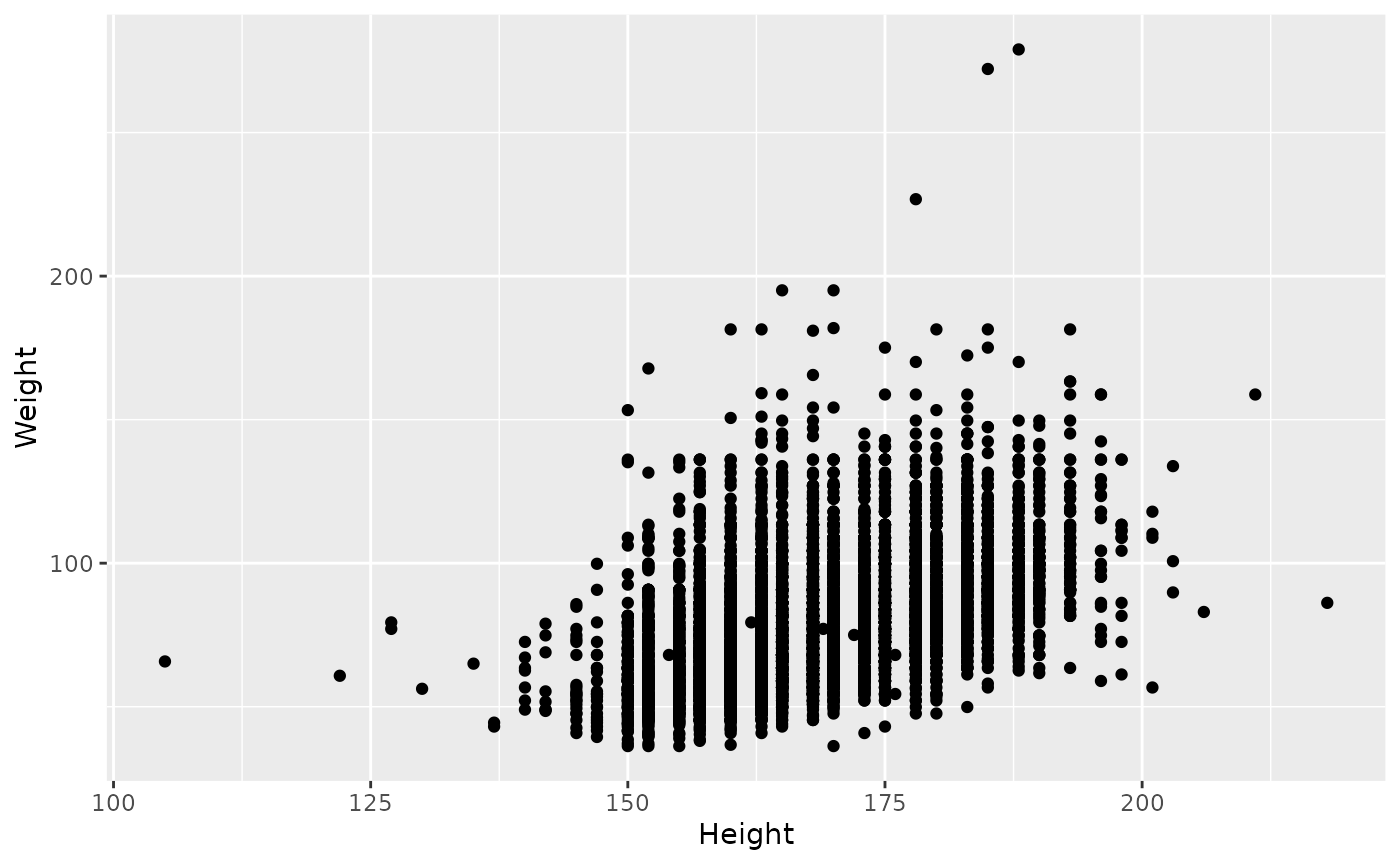
Yes there looks like a relationship. Add
geom_smooth(method = "lm") to fit a smoothed relationship
to the points; method = "lm" indicates that the smoothed
line should be a linear regression.
ggplot(brfss_2010) +
aes(x = Height, y = Weight) +
geom_point() +
geom_smooth(method = "lm")
## `geom_smooth()` using formula = 'y ~ x'
## Warning: Removed 450 rows containing non-finite outside the scale range
## (`stat_smooth()`).
## Warning: Removed 450 rows containing missing values or values outside the scale range
## (`geom_point()`).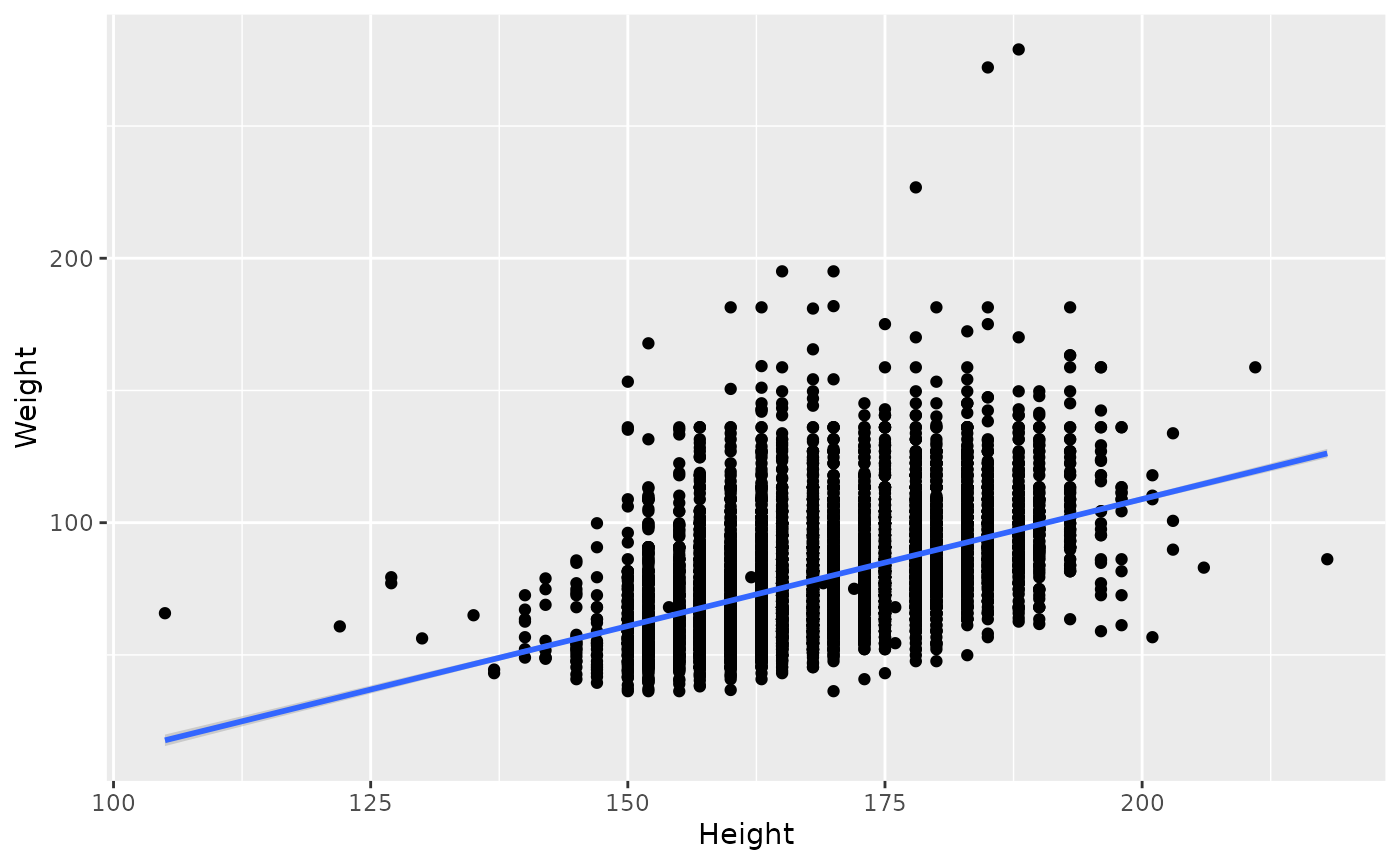
The relationship between height and weight likely depends on sex. Add
color = Sex to the aes() argument, so each
geom (both points and smoothed line) is colored by Sex.
ggplot(brfss_2010) +
aes(x = Height, y = Weight, color = Sex) +
geom_point() +
geom_smooth(method = "lm")
## `geom_smooth()` using formula = 'y ~ x'
## Warning: Removed 450 rows containing non-finite outside the scale range
## (`stat_smooth()`).
## Warning: Removed 450 rows containing missing values or values outside the scale range
## (`geom_point()`).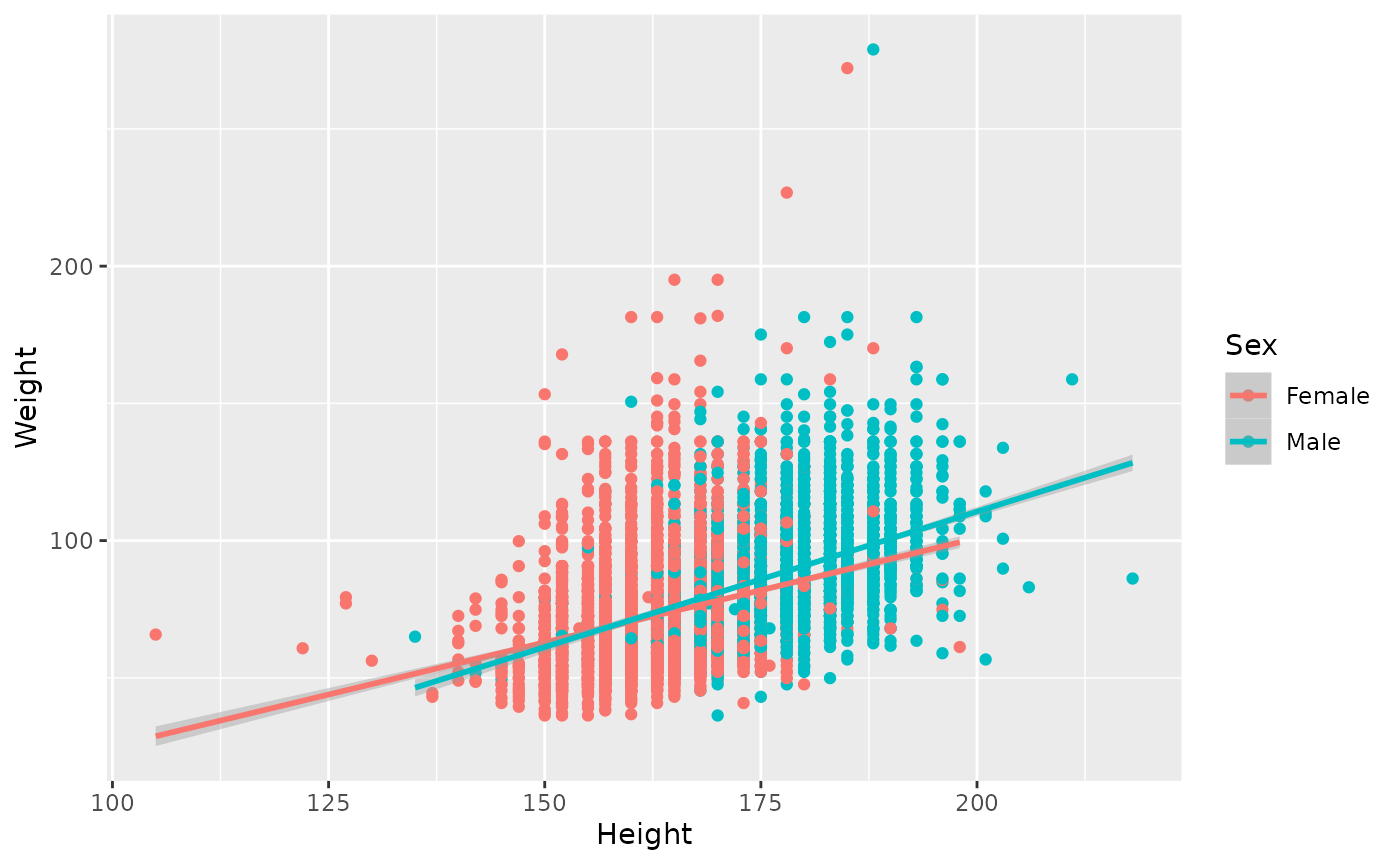
The lines cover the range of values for each sex; the relationship between height and weight appears slightly steeper for males than females.
Has the relationship between height and weight of males changed
between 1990 and 2010? Change the dataset to brfss_male,
color by Year, and add a title to the plot so that we know
a little bit more about the relationship. Also, explore a data
transformation by using the log of weight
ggplot(brfss_male) +
aes(x = Height, y = log10(Weight), color = Year) +
geom_point() +
geom_smooth(method = "lm") +
labs(title = "BRFSS Male Subset")
## `geom_smooth()` using formula = 'y ~ x'
## Warning: Removed 104 rows containing non-finite outside the scale range
## (`stat_smooth()`).
## Warning: Removed 104 rows containing missing values or values outside the scale range
## (`geom_point()`).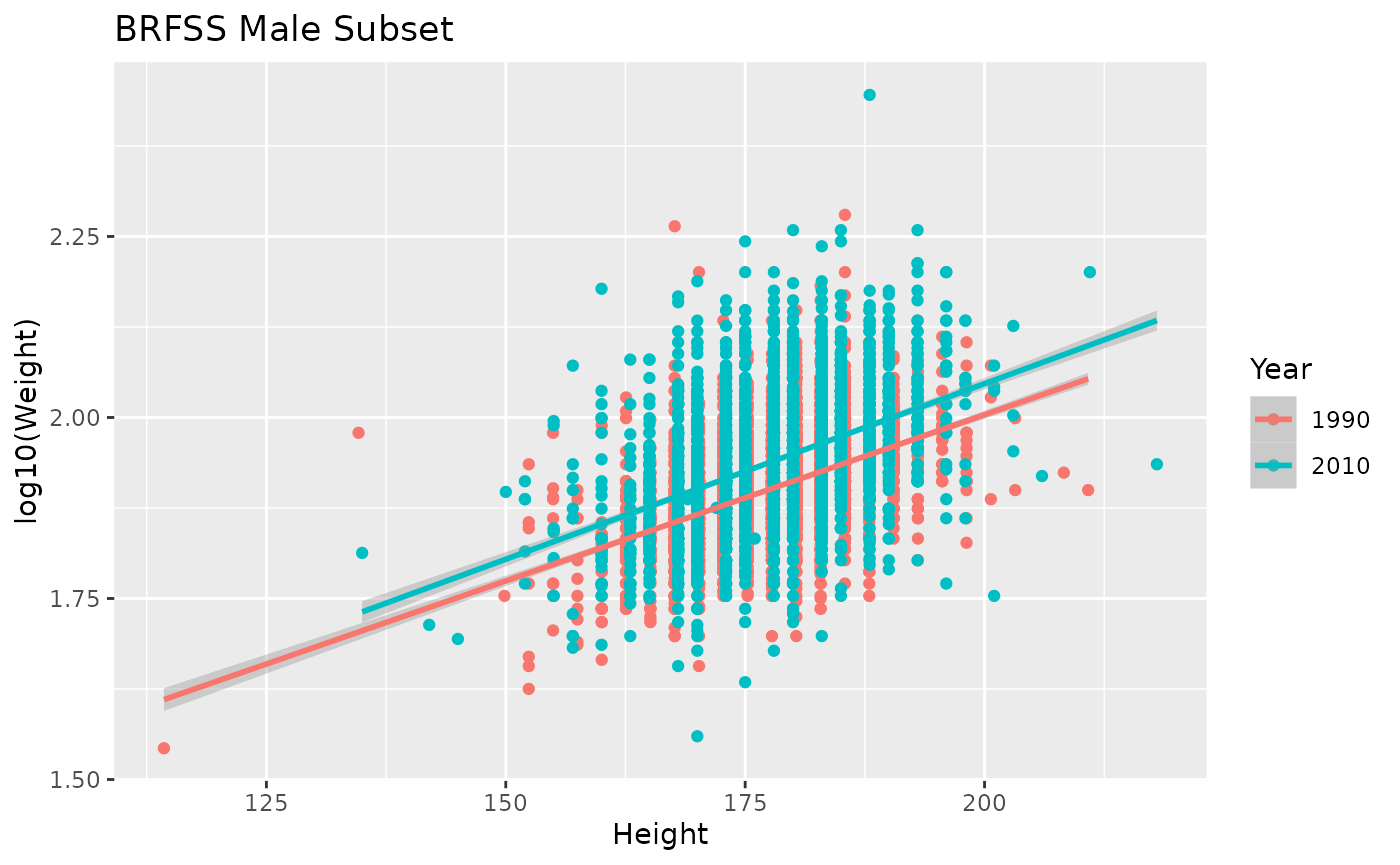
The figure suggests that 2010 males are heavier than 1990 males at all heights, and hints that the relationship between weight and height is steeper in 2010; a formal statistical analysis is required for further confidence.
Statistical analysis
This section illustrates basic statistical tests; it would be interesting to sit down with a statistician to discuss the subtleties of a more correct analysis.
We will explroe base R functions
-
t.test()for performing two-sample t-tests. -
wilcox.test(), a non-parametric two-sample test. -
lm()to fit a linear model (regression). -
summary()to summarize the result of the linear model as an ANOVA table with R-squared measures summarizing goodness of fit.
Difference between groups
Here is a partial summary of the brfss_2010 data
subset
brfss_2010 |>
group_by(Sex) |>
summarize(
n = n(),
ave_age = mean(Age, na.rm = TRUE),
ave_wt = mean(Weight, na.rm = TRUE),
ave_ht = mean(Height, na.rm = TRUE)
)
## # A tibble: 2 × 5
## Sex n ave_age ave_wt ave_ht
## <fct> <int> <dbl> <dbl> <dbl>
## 1 Female 6321 57.1 73.0 163.
## 2 Male 3679 56.2 88.8 178.Is there statistical support for the small difference between average ages of Female and Male respondents?
Use t.test() to compare two groups. After studying the
help page ?t.test, we use the ‘formula’ notation to test
for differences in Age as a function of Sex, Age ~ Sex,
using the subset brfss_2010 as the data source.
t.test(Age ~ Sex, brfss_2010)
##
## Welch Two Sample t-test
##
## data: Age by Sex
## t = 2.4497, df = 7768.7, p-value = 0.01432
## alternative hypothesis: true difference in means between group Female and group Male is not equal to 0
## 95 percent confidence interval:
## 0.1674909 1.5091167
## sample estimates:
## mean in group Female mean in group Male
## 57.08824 56.24993The summary reports (at the bottom) mean ages of Male and Female respondents consistent with our own calculations, so we know we have not made some kind of serious blunder in formulating the test. The t-statistic of 2.4497162, with P-value 0.0143188 is significant.
What about differences in Male Weight between 1990 and 2010?
t.test(Weight ~ Year, brfss_male)
##
## Welch Two Sample t-test
##
## data: Weight by Year
## t = -20.751, df = 6549.4, p-value < 2.2e-16
## alternative hypothesis: true difference in means between group 1990 and group 2010 is not equal to 0
## 95 percent confidence interval:
## -8.390845 -6.942327
## sample estimates:
## mean in group 1990 mean in group 2010
## 81.17999 88.84657Conversation with a statistician might make us concerned about whether assumptions of the t-test are fully satisfied, e.g., the data are supposed to be normally distributed, but as we saw in the box plots weights are skewed. We could try to transform the data (formal approaches to assess appropriateness of data transformations are available), e.g., by using log Weight
t.test(log10(Weight) ~ Year, brfss_male)
##
## Welch Two Sample t-test
##
## data: log10(Weight) by Year
## t = -20.202, df = 6985.4, p-value < 2.2e-16
## alternative hypothesis: true difference in means between group 1990 and group 2010 is not equal to 0
## 95 percent confidence interval:
## -0.03971348 -0.03268805
## sample estimates:
## mean in group 1990 mean in group 2010
## 1.903711 1.939912Or we might use a statistic like the Wilcoxon test that makes fewer assumptions about the underlying statistical distribution.
wilcox.test(Weight ~ Year, brfss_male)
##
## Wilcoxon rank sum test with continuity correction
##
## data: Weight by Year
## W = 5754766, p-value < 2.2e-16
## alternative hypothesis: true location shift is not equal to 0Regardless of the details of the analysis, the difference in Male Weight between 1990 and 2010 is highly significant.
Linear regression
The ‘BRFSS Male Subset’ figure shows linear relations between the square root of Weight and Height in each year.
ggplot(brfss_male) +
aes(x = Height, y = log10(Weight), color = Year) +
geom_point() +
geom_smooth(method = "lm") +
labs(title = "BRFSS Male Subset")
## `geom_smooth()` using formula = 'y ~ x'
## Warning: Removed 104 rows containing non-finite outside the scale range
## (`stat_smooth()`).
## Warning: Removed 104 rows containing missing values or values outside the scale range
## (`geom_point()`).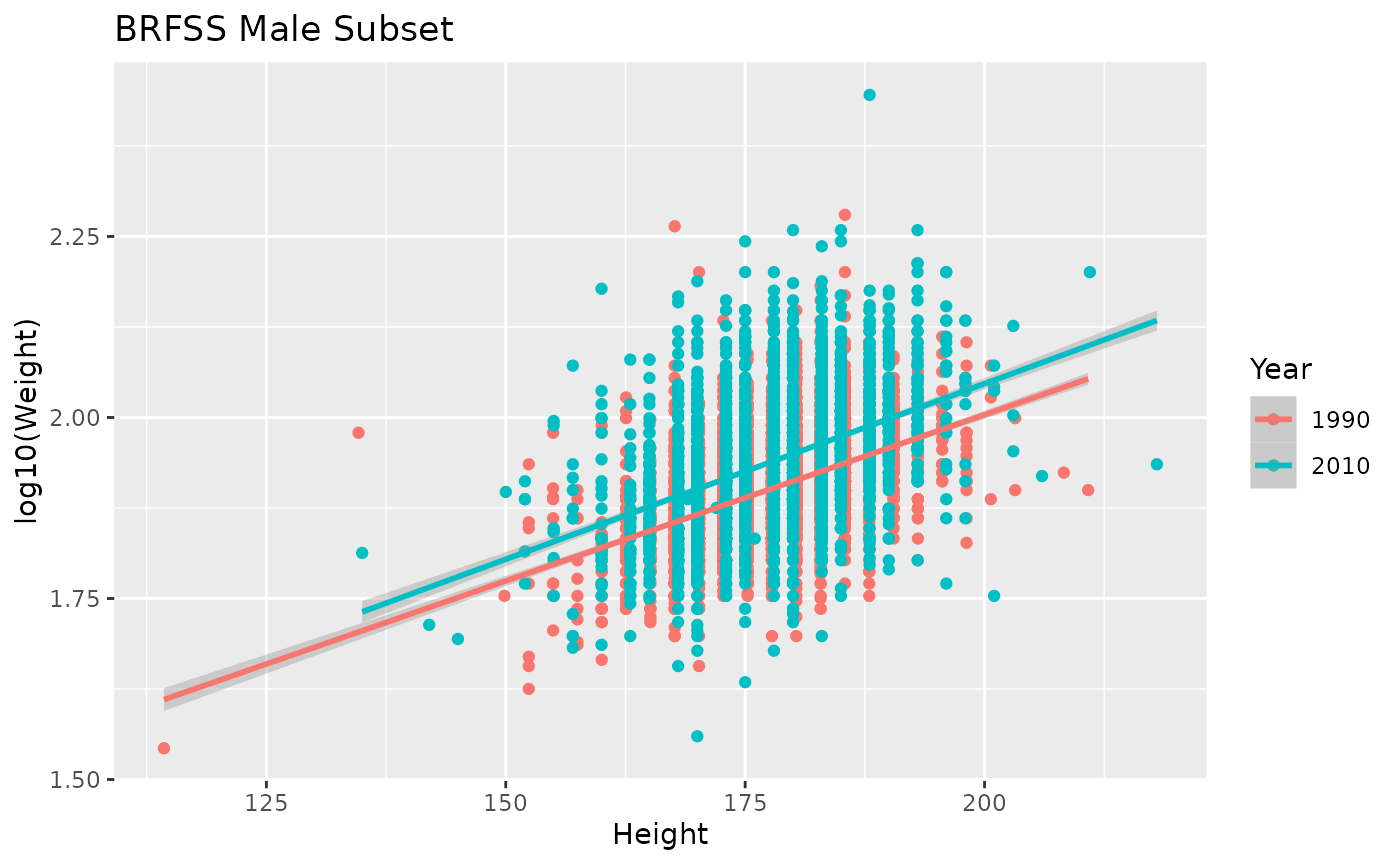
How would we calculate this regression, and assess its significance? We focus on Male 2010 data.
brfss_male_2010 <-
brfss_male |>
filter(Year == "2010")The answer is to fit a linear regression to the data. In R,
this is done by fitting a linear model (lm()) and then
summarizing the result.
## fit the linear model
fit <- lm(log10(Weight) ~ Height, brfss_male_2010)
## summarize the fit, including a statistical assessment of the fit
summary(fit)
##
## Call:
## lm(formula = log10(Weight) ~ Height, data = brfss_male_2010)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.34142 -0.05229 -0.00645 0.04462 0.45706
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 1.0767107 0.0309565 34.78 <2e-16 ***
## Height 0.0048498 0.0001737 27.91 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.07824 on 3617 degrees of freedom
## (60 observations deleted due to missingness)
## Multiple R-squared: 0.1772, Adjusted R-squared: 0.177
## F-statistic: 779.2 on 1 and 3617 DF, p-value: < 2.2e-16The ANOVA table shows that the relationship is highly significant. The ‘Adjusted R-squared’ value indicates that about 17% of the variation in Weight is accounted for by Height. The estimated coefficient associated with Height is the slope of the line, indicating that the square root of Weight increases by about 0.0048498 for every increase in Height of 1 cm.
As an aside, one might hope that plot(fit) would plot
the regression line. Actually, it creates a series of diagnostic plots
that help us assess the appropriateness of our choice of a linear model
for describing this data.
More advanced analysis using R to test, e.g., for differences in the intercept or slope of the regression in 1990 versus 2010 are straight-forward to implement, but require more sophisticated statistical understanding.
Acute Lymphocytic Leukemia (ALL)
This data set is from an old microarray experiment investigating acute lymphocytic leukemia (ALL) (PMID 14684422, 16243790; the data has been extracted from the ALL Bioconductor package). We focus on phenotypes of 128 patients.
We use many of the sample dplyr functions as in the BRFSS data set, but encounter a number of new R functions useful for specific tasks.
-
ifelse()to compose a new vector based on values in a logical vector. -
startsWith()to test whether each element of a character vector starts with a particular prefix. -
%in%, used aslhs %in% rhsasks whether each element in the vectorlhsis a member of the vectorrhs.
Data input
Use readr::read_csv() to input the ‘ALL.csv’ file.
all_file <- file.choose() # look for 'Rpg520/week-04/extdata/all.csv'
all <- readr::read_csv(all_file)
glimpse(all)
## Rows: 128
## Columns: 21
## $ cod <chr> "1005", "1010", "3002", "4006", "4007", "4008", "4010…
## $ diagnosis <chr> "5/21/1997", "3/29/2000", "6/24/1998", "7/17/1997", "…
## $ sex <chr> "M", "M", "F", "M", "M", "M", "F", "M", "M", "M", "M"…
## $ age <dbl> 53, 19, 52, 38, 57, 17, 18, 16, 15, 40, 33, 55, 5, 18…
## $ BT <chr> "B2", "B2", "B4", "B1", "B2", "B1", "B1", "B1", "B2",…
## $ remission <chr> "CR", "CR", "CR", "CR", "CR", "CR", "CR", "CR", "CR",…
## $ CR <chr> "CR", "CR", "CR", "CR", "CR", "CR", "CR", "CR", "CR",…
## $ date.cr <chr> "8/6/1997", "6/27/2000", "8/17/1998", "9/8/1997", "9/…
## $ `t(4;11)` <lgl> FALSE, FALSE, NA, TRUE, FALSE, FALSE, FALSE, FALSE, F…
## $ `t(9;22)` <lgl> TRUE, FALSE, NA, FALSE, FALSE, FALSE, FALSE, FALSE, F…
## $ cyto.normal <lgl> FALSE, FALSE, NA, FALSE, FALSE, FALSE, FALSE, FALSE, …
## $ citog <chr> "t(9;22)", "simple alt.", NA, "t(4;11)", "del(6q)", "…
## $ mol.biol <chr> "BCR/ABL", "NEG", "BCR/ABL", "ALL1/AF4", "NEG", "NEG"…
## $ `fusion protein` <chr> "p210", NA, "p190", NA, NA, NA, NA, NA, NA, "p190", "…
## $ mdr <chr> "NEG", "POS", "NEG", "NEG", "NEG", "NEG", "POS", "NEG…
## $ kinet <chr> "dyploid", "dyploid", "dyploid", "dyploid", "dyploid"…
## $ ccr <lgl> FALSE, FALSE, FALSE, FALSE, FALSE, FALSE, FALSE, FALS…
## $ relapse <lgl> FALSE, TRUE, TRUE, TRUE, TRUE, TRUE, TRUE, TRUE, TRUE…
## $ transplant <lgl> TRUE, FALSE, FALSE, FALSE, FALSE, FALSE, FALSE, FALSE…
## $ f.u <chr> "BMT / DEATH IN CR", "REL", "REL", "REL", "REL", "REL…
## $ `date last seen` <chr> NA, "8/28/2000", "10/15/1999", "1/23/1998", "11/4/199…Exploration & cleaning
For simplicity we focus on the following columns:
- ‘cod’ is a unique identifier
- ‘sex’. We will recode this to a factor with levels ‘Female’ and ‘Male’, so that there is no ambiguity about its meaning.
- ‘age’.
- ‘BT’ represents B- or T- cell status. It is too fine-grained, so we will recode this to a new variable ‘BorT’.
- ‘mol.biol’ summarizes status with respect to chromosomal status. We will filter the data to contain only patients with values ‘BCR/ABL’ (the classic BCR / ABL inversion) or ‘NEG’ (no chromosomal changes). We will recode the result as a factor with levels ‘BCR/ABL’ and ‘NEG’.
select()
Use select() to select just the columns of interest.
Re-assign the result to all.
all <-
all |>
select(cod, sex, age, BT, mol.biol)
ifelse() and factor()
‘sex’ is currently a character vector with two values ‘F’ and ‘M’, as well as missing values
all |>
count(sex)
## # A tibble: 3 × 2
## sex n
## <chr> <int>
## 1 F 42
## 2 M 83
## 3 NA 3We can remove individuals whose sex is unknown using
filter(!is.na(sex)). Use ifelse() to translate
‘F’ and ‘M’ to ‘Female’ and ‘Male’, and then factor() so
that this column is a factor
all |>
filter(!is.na(sex)) |>
mutate(
sex = factor(
ifelse(sex == "F", "Female", "Male"),
levels = c("Female", "Male")
)
)
## # A tibble: 125 × 5
## cod sex age BT mol.biol
## <chr> <fct> <dbl> <chr> <chr>
## 1 1005 Male 53 B2 BCR/ABL
## 2 1010 Male 19 B2 NEG
## 3 3002 Female 52 B4 BCR/ABL
## 4 4006 Male 38 B1 ALL1/AF4
## 5 4007 Male 57 B2 NEG
## 6 4008 Male 17 B1 NEG
## 7 4010 Female 18 B1 NEG
## 8 4016 Male 16 B1 NEG
## 9 6002 Male 15 B2 NEG
## 10 8001 Male 40 B2 BCR/ABL
## # ℹ 115 more rows
startsWith()
The BT column describes whether individuals have B- or
T-cell ALL. There are several different grades of each.
all |>
count(BT)
## # A tibble: 10 × 2
## BT n
## <chr> <int>
## 1 B 5
## 2 B1 19
## 3 B2 36
## 4 B3 23
## 5 B4 12
## 6 T 5
## 7 T1 1
## 8 T2 15
## 9 T3 10
## 10 T4 2We are interested in simplifying this to just two values, ‘B’ or ‘T’,
based on the first letter of each vector element. The
startsWith() function returns TRUE each time
an element of the first argument starts with the second argument.
x <- c("B", "B1", "T")
startsWith(x, "B")
## [1] TRUE TRUE FALSEWe can combine this with ifelse() and
factor() to mutate BT
all |>
mutate(
BT = factor(
ifelse(startsWith(BT, "B"), "B", "T"),
levels = c("B", "T")
)
)
## # A tibble: 128 × 5
## cod sex age BT mol.biol
## <chr> <chr> <dbl> <fct> <chr>
## 1 1005 M 53 B BCR/ABL
## 2 1010 M 19 B NEG
## 3 3002 F 52 B BCR/ABL
## 4 4006 M 38 B ALL1/AF4
## 5 4007 M 57 B NEG
## 6 4008 M 17 B NEG
## 7 4010 F 18 B NEG
## 8 4016 M 16 B NEG
## 9 6002 M 15 B NEG
## 10 8001 M 40 B BCR/ABL
## # ℹ 118 more rows
%in%
‘mol.biol’ is also a character vector with several different values.
all |>
count(mol.biol)
## # A tibble: 6 × 2
## mol.biol n
## <chr> <int>
## 1 ALL1/AF4 10
## 2 BCR/ABL 37
## 3 E2A/PBX1 5
## 4 NEG 74
## 5 NUP-98 1
## 6 p15/p16 1We will filter all to only include ‘BCR/ABL’ or ‘NEG’
samples using %in%. %in% is an ‘infix’
operator (like ~) with a left-hand side and a right-hand
side. The left-hand side is a vector of values that we are interested
in. The right-hand side is also a vector, and represents a
(mathematical) ‘set’ of values. %in% asks, for each element
of the left-hand size, if the element is a member of the set on the
right hand side.
Filtering the all dataset is accomplished with
all |>
filter(mol.biol %in% c("BCR/ABL", "NEG"))
## # A tibble: 111 × 5
## cod sex age BT mol.biol
## <chr> <chr> <dbl> <chr> <chr>
## 1 1005 M 53 B2 BCR/ABL
## 2 1010 M 19 B2 NEG
## 3 3002 F 52 B4 BCR/ABL
## 4 4007 M 57 B2 NEG
## 5 4008 M 17 B1 NEG
## 6 4010 F 18 B1 NEG
## 7 4016 M 16 B1 NEG
## 8 6002 M 15 B2 NEG
## 9 8001 M 40 B2 BCR/ABL
## 10 8011 M 33 B3 BCR/ABL
## # ℹ 101 more rowsRe-coding the filtered values of ‘mol.biol’ uses
factor() in a way that we have already seen with ‘sex’.
Data cleaning pipeline
Putting these filtering and re-coding steps together, our initial cleaning results in
all_subset <-
all |>
## columns of interest
select(cod, sex, age, BT, mol.biol) |>
## rows of interest
filter(
!is.na(sex),
mol.biol %in% c("BCR/ABL", "NEG")
) |>
## re-coding sex, mol.bio, BT
mutate(
sex = factor(
ifelse(sex == "F", "Female", "Male"),
levels = c("Female", "Male")
),
BT = factor(
ifelse(startsWith(BT, "B"), "B", "T"),
levels = c("B", "T")
),
mol.biol = factor(mol.biol, levels = c("BCR/ABL", "NEG"))
)Data exploration
A basic characterize of our subset might group by sex and summarize the number and average age of each group.
all_subset |>
group_by(sex) |>
summarize(
n = n(),
av_age = mean(age, na.rm = TRUE)
)
## # A tibble: 2 × 3
## sex n av_age
## <fct> <int> <dbl>
## 1 Female 35 34.8
## 2 Male 74 30.9There are about twice as many Male as Female samples; the average age of Female samples is about 4 years older than Male samples.
We can visualize the distribution of ages in several ways, as
illustrated in the previous case study. Here we create a box plot with
geom_boxplot(), then overlay the individual points using
geom_jitter() (this geom displays each point but with a bit
of ‘jitter’ away from its actual value – instead of being plotted
exactly on the ‘Female’ or ‘Male’ line, the points are offset a random
amount).
ggplot(all_subset) +
aes(x = sex, y = age) +
geom_boxplot() +
geom_jitter(width = .2)
## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_boxplot()`).
## Warning: Removed 2 rows containing missing values or values outside the scale range
## (`geom_point()`).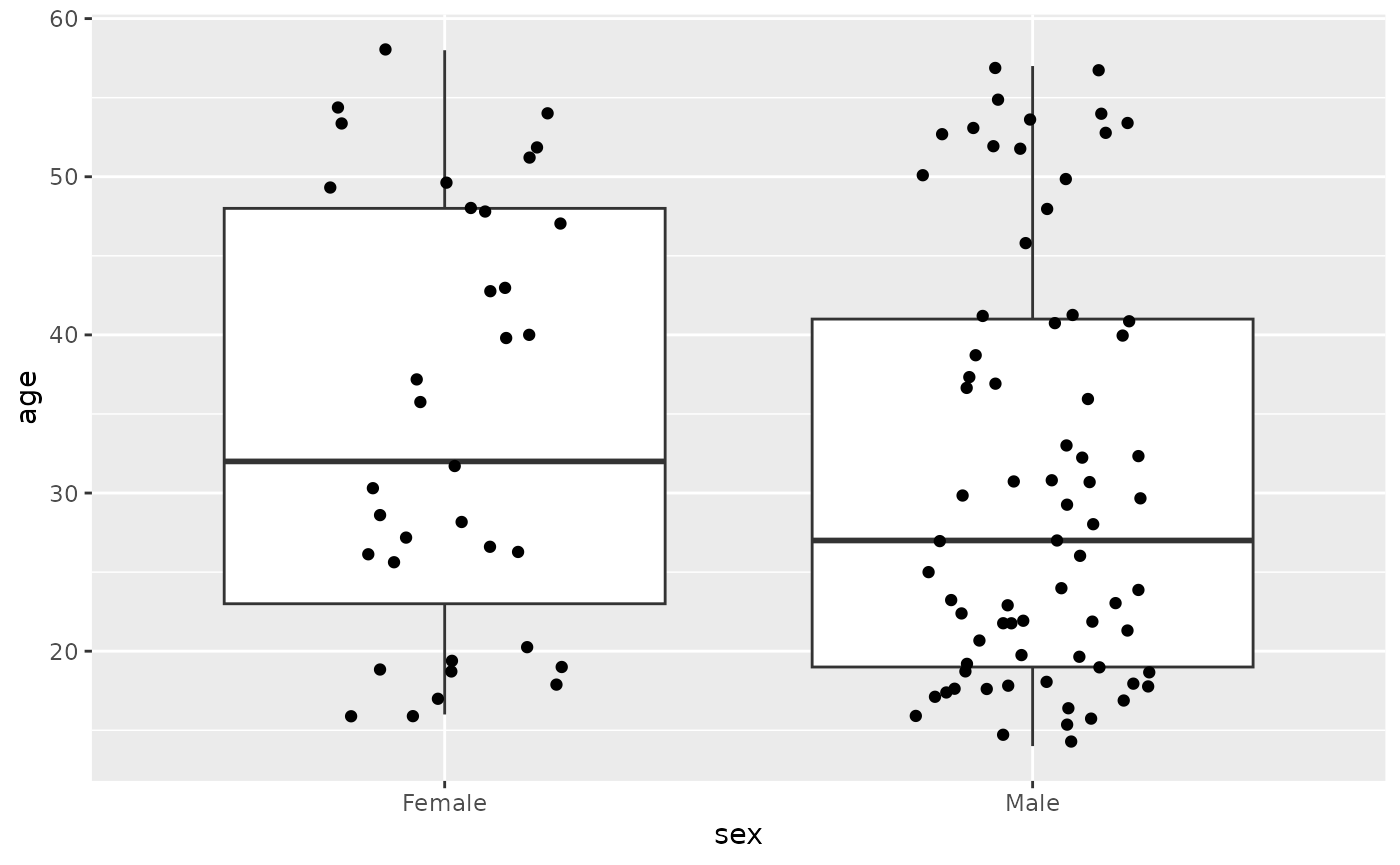
The following table counts the number of ‘BCR/ABL’ and ‘NEG’ among ‘Female’ and ‘Male’ samples; it seems like there is a disproportionate number of ‘Male’ ‘NEG’ (chromosomally normal) individuals.
all_subset |>
count(mol.biol, sex) |>
tidyr::pivot_wider(names_from = "sex", values_from = "n")
## # A tibble: 2 × 3
## mol.biol Female Male
## <fct> <int> <int>
## 1 BCR/ABL 16 21
## 2 NEG 19 53Here is a similar tally of B- and T-cell ALL for ‘Female’ and ‘Male’ samples
all_subset |>
count(BT, sex) |>
tidyr::pivot_wider(names_from = "sex", values_from = "n")
## # A tibble: 2 × 3
## BT Female Male
## <fct> <int> <int>
## 1 B 28 50
## 2 T 7 24… and a count of ‘BT’ and ‘mol.biol’ by sex
all_subset |>
count(BT, mol.biol, sex) |>
tidyr::pivot_wider(names_from = "sex", values_from = "n")
## # A tibble: 3 × 4
## BT mol.biol Female Male
## <fct> <fct> <int> <int>
## 1 B BCR/ABL 16 21
## 2 B NEG 12 29
## 3 T NEG 7 24Note that there are not T-cell, BCR/ABL samples. Is this for biological reasons, or is an artifact of the procedures used in data collection? Does it limit or bias the statistical questions that can be asked?
Statistical analysis
A t-test indicates that the difference in Male and Female age noted above is not statistically significant
t.test(age ~ sex, all_subset)
##
## Welch Two Sample t-test
##
## data: age by sex
## t = 1.3901, df = 66.316, p-value = 0.1691
## alternative hypothesis: true difference in means between group Female and group Male is not equal to 0
## 95 percent confidence interval:
## -1.687236 9.424537
## sample estimates:
## mean in group Female mean in group Male
## 34.77143 30.90278We noted differences in the number ‘BCR/ABL’ and ‘NEG’ samples with
respect to sex. A Chi-squared test can be used to assess
whether these differences are statistically significant. Look at the
help page ?chisq.test. The idea is that the first argument
is a vector of observations of one variable (e.g.,
mol.biol) and the second argument is the second variable
(sex). Unlike t.test, there is no
data= argument, and no formula (~) interface.
In base R one might use $ to access individual
columns of a tibble, so the test could be performed with
chisq.test(all_subset$mol.biol, all_subset$sex)A convenient variation of this is to use the function
with(), which allows one to write the test as
all_subset |>
with(chisq.test(mol.biol, sex))
##
## Pearson's Chi-squared test with Yates' continuity correction
##
## data: mol.biol and sex
## X-squared = 2.4586, df = 1, p-value = 0.1169The P-value indicates that the differences in counts of ‘BCR/ABL’ and
‘NEG’ observed between ‘Female’ and ‘Male’ are not statistically
supported. A similar conclusion applies with BT and
sex.
all_subset |>
with(chisq.test(BT, sex))
##
## Pearson's Chi-squared test with Yates' continuity correction
##
## data: BT and sex
## X-squared = 1.2454, df = 1, p-value = 0.2644Session information
For reproducibility, I record the software versions used to create this document
sessionInfo()
## R version 4.3.3 (2024-02-29)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggplot2_3.5.0 dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] sass_0.4.8 utf8_1.2.4 generics_0.1.3 tidyr_1.3.1
## [5] lattice_0.22-5 stringi_1.8.3 hms_1.1.3 digest_0.6.34
## [9] magrittr_2.0.3 evaluate_0.23 grid_4.3.3 fastmap_1.1.1
## [13] Matrix_1.6-5 jsonlite_1.8.8 mgcv_1.9-1 purrr_1.0.2
## [17] fansi_1.0.6 scales_1.3.0 textshaping_0.3.7 jquerylib_0.1.4
## [21] cli_3.6.2 rlang_1.1.3 crayon_1.5.2 splines_4.3.3
## [25] bit64_4.0.5 munsell_0.5.0 withr_3.0.0 cachem_1.0.8
## [29] yaml_2.3.8 tools_4.3.3 parallel_4.3.3 tzdb_0.4.0
## [33] memoise_2.0.1 colorspace_2.1-0 vctrs_0.6.5 R6_2.5.1
## [37] lifecycle_1.0.4 stringr_1.5.1 fs_1.6.3 bit_4.0.5
## [41] vroom_1.6.5 ragg_1.2.7 pkgconfig_2.0.3 desc_1.4.3
## [45] pkgdown_2.0.7 pillar_1.9.0 bslib_0.6.1 gtable_0.3.4
## [49] glue_1.7.0 systemfonts_1.0.5 highr_0.10 xfun_0.42
## [53] tibble_3.2.1 tidyselect_1.2.0 knitr_1.45 farver_2.1.1
## [57] nlme_3.1-164 htmltools_0.5.7 labeling_0.4.3 rmarkdown_2.25
## [61] readr_2.1.5 compiler_4.3.3