Log on to https://ondemand.ccr.buffalo.edu
-
Set up RStudio for today
source('/projects/rpci/rplect/Rpg520/Rsetdirs.R') -
Let RStudio know about your new working directory
Introduction
Today we work through two more cancer-relevant case studies. The first looks at single-cell RNAseq, aiming to understand how we can visualize a ‘UMAP’ projection of cell types. The second introduces survival analysis and working with data to present a ‘Kaplan-Meier’ curve. The workshop material is by no means exhaustive!
Single-cell RNAseq
This case study illustrates how R can be used to understand single-cell RNAseq data, assuming that the ‘heavy lifting’ of transforming raw FASTQ files to normalized matrices of counts measuring expression of each gene in each cell has been done by, e.g., a bioinformatics core. R can be used to summarize biological sample and cell attributes, e.g., the number of donors and cells per donor. Visualizations like UMAP plots showing cell expression patterns in two-dimensional space can be easily generated and annotated. Individual genes can be annotated with additional information, e.g., with a description of the gene or of the genes in particular pathways. The next article introduces more comprehensive work flows.
The data set come from the CELLxGENE data portal. The dataset is of breast epithelial cells. The dataset is relatively typical of single-cell experimental data sets likely to be generated in individual research projects. The data were downloaded using the cellxgenedp pacakge, with information extracted from the file using the zellkonverter and rhdf5 packages. All of these packages are available through Bioconductor, and have vignettes describing their use.
The main outcome of this case study is an interactive scatter plot; most of the data input, cleaning and visualization steps are similar to Day 2.
Input
As before, we use dplyr for data manipulation, and readr for data input.
Read a ‘csv’ file summarizing infomration about each cell in the experiment.
cell_file <- file.choose() # find 'scrnaseq-cell-data.csv'
cell <- readr::read_csv(cell_file)
glimpse(cell)
## Rows: 31,696
## Columns: 37
## $ cell_id <chr> "CMGpool_AAACCCAAGGACAACC", "…
## $ UMAP_1 <dbl> -5.9931696, -6.7217583, -9.01…
## $ UMAP_2 <dbl> -1.9311870, -1.3059803, -3.39…
## $ donor_id <chr> "pooled [D9,D7,D8,D10,D6]", "…
## $ self_reported_ethnicity_ontology_term_id <chr> "HANCESTRO:0005", "HANCESTRO:…
## $ donor_BMI <chr> "pooled [30.5,22.7,23.5,26.8,…
## $ donor_times_pregnant <chr> "pooled [3,0,3,2,2]", "pooled…
## $ family_history_breast_cancer <chr> "pooled [unknown,False,False,…
## $ organism_ontology_term_id <chr> "NCBITaxon:9606", "NCBITaxon:…
## $ tyrer_cuzick_lifetime_risk <chr> "pooled [12,14.8,8.8,14.3,20.…
## $ sample_uuid <chr> "pooled [f008c67a-abb4-4563-8…
## $ sample_preservation_method <chr> "cryopreservation", "cryopres…
## $ tissue_ontology_term_id <chr> "UBERON:0035328", "UBERON:003…
## $ development_stage_ontology_term_id <chr> "HsapDv:0000087", "HsapDv:000…
## $ suspension_uuid <chr> "38d793cb-d811-4863-aec0-2fa7…
## $ suspension_type <chr> "cell", "cell", "cell", "cell…
## $ library_uuid <chr> "385d8d7c-5038-4f0e-b7f3-ec9a…
## $ assay_ontology_term_id <chr> "EFO:0009922", "EFO:0009922",…
## $ mapped_reference_annotation <chr> "GENCODE 28", "GENCODE 28", "…
## $ is_primary_data <lgl> TRUE, TRUE, TRUE, TRUE, TRUE,…
## $ cell_type_ontology_term_id <chr> "CL:0011026", "CL:0011026", "…
## $ author_cell_type <chr> "luminal progenitor", "lumina…
## $ disease_ontology_term_id <chr> "PATO:0000461", "PATO:0000461…
## $ sex_ontology_term_id <chr> "PATO:0000383", "PATO:0000383…
## $ nCount_RNA <dbl> 2937, 5495, 5598, 3775, 2146,…
## $ nFeature_RNA <dbl> 1183, 1827, 2037, 1448, 1027,…
## $ percent.mito <dbl> 0.02076949, 0.03676069, 0.043…
## $ seurat_clusters <dbl> 3, 3, 24, 1, 3, 1, 1, 4, 1, 0…
## $ sample_id <chr> "CMGpool", "CMGpool", "CMGpoo…
## $ cell_type <chr> "progenitor cell", "progenito…
## $ assay <chr> "10x 3' v3", "10x 3' v3", "10…
## $ disease <chr> "normal", "normal", "normal",…
## $ organism <chr> "Homo sapiens", "Homo sapiens…
## $ sex <chr> "female", "female", "female",…
## $ tissue <chr> "upper outer quadrant of brea…
## $ self_reported_ethnicity <chr> "European", "European", "Euro…
## $ development_stage <chr> "human adult stage", "human a…Exploration & cleaning
Summarize information – how many donors, what developmental stage, what ethnicity?
cell |>
count(donor_id, development_stage, self_reported_ethnicity)
## # A tibble: 7 × 4
## donor_id development_stage self_reported_ethnicity n
## <chr> <chr> <chr> <int>
## 1 D1 35-year-old human stage European 2303
## 2 D11 43-year-old human stage Chinese 7454
## 3 D2 60-year-old human stage European 864
## 4 D3 44-year-old human stage African American 2517
## 5 D4 42-year-old human stage European 1771
## 6 D5 21-year-old human stage European 2244
## 7 pooled [D9,D7,D8,D10,D6] human adult stage European 14543What cell types have been annotated?
cell |>
count(cell_type)
## # A tibble: 6 × 2
## cell_type n
## <chr> <int>
## 1 B cell 215
## 2 basal cell 7040
## 3 endocrine cell 64
## 4 endothelial cell of lymphatic vessel 133
## 5 luminal epithelial cell of mammary gland 4257
## 6 progenitor cell 19987Cell types for each ethnicity?
cell |>
count(self_reported_ethnicity, cell_type) |>
tidyr::pivot_wider(
names_from = "self_reported_ethnicity",
values_from = "n"
)
## # A tibble: 6 × 4
## cell_type `African American` Chinese European
## <chr> <int> <int> <int>
## 1 B cell 5 73 137
## 2 basal cell 583 3367 3090
## 3 endothelial cell of lymphatic vessel 11 31 91
## 4 luminal epithelial cell of mammary gland 809 187 3261
## 5 progenitor cell 1109 3755 15123
## 6 endocrine cell NA 41 23Reflecting on this – there is no replication across non-European ethnicity, so no statistical insights available. Pooled samples probably require careful treatment in any downstream analysis.
UMAP visualization
In a single-cell experiment, each cell has been characterized for
gene expression at 1000’s of genes, so a cell can be though of as
occupying a position in 1000’s of dimensions. UMAP (‘Uniform Manifold
Approximation and Projection’) is a method for reducing the
dimensionality of the space the cell occupies, to simplify visualization
and other operations. The UMAP_1 and UMAP_2
columns contain the coordinates of each cell in the space defined by
UMAP applied to the cell expression values.
Use the ‘UMAP’ columns to visualize gene expression. This code
illustrates that ggplot() + ... actually returns an
object that can be assigned to a variable (plt) to
be used in subsequent computations.
library(ggplot2)
plt <-
ggplot(cell) +
aes(UMAP_1, UMAP_2, color = cell_type) +
geom_point(pch = ".")
plt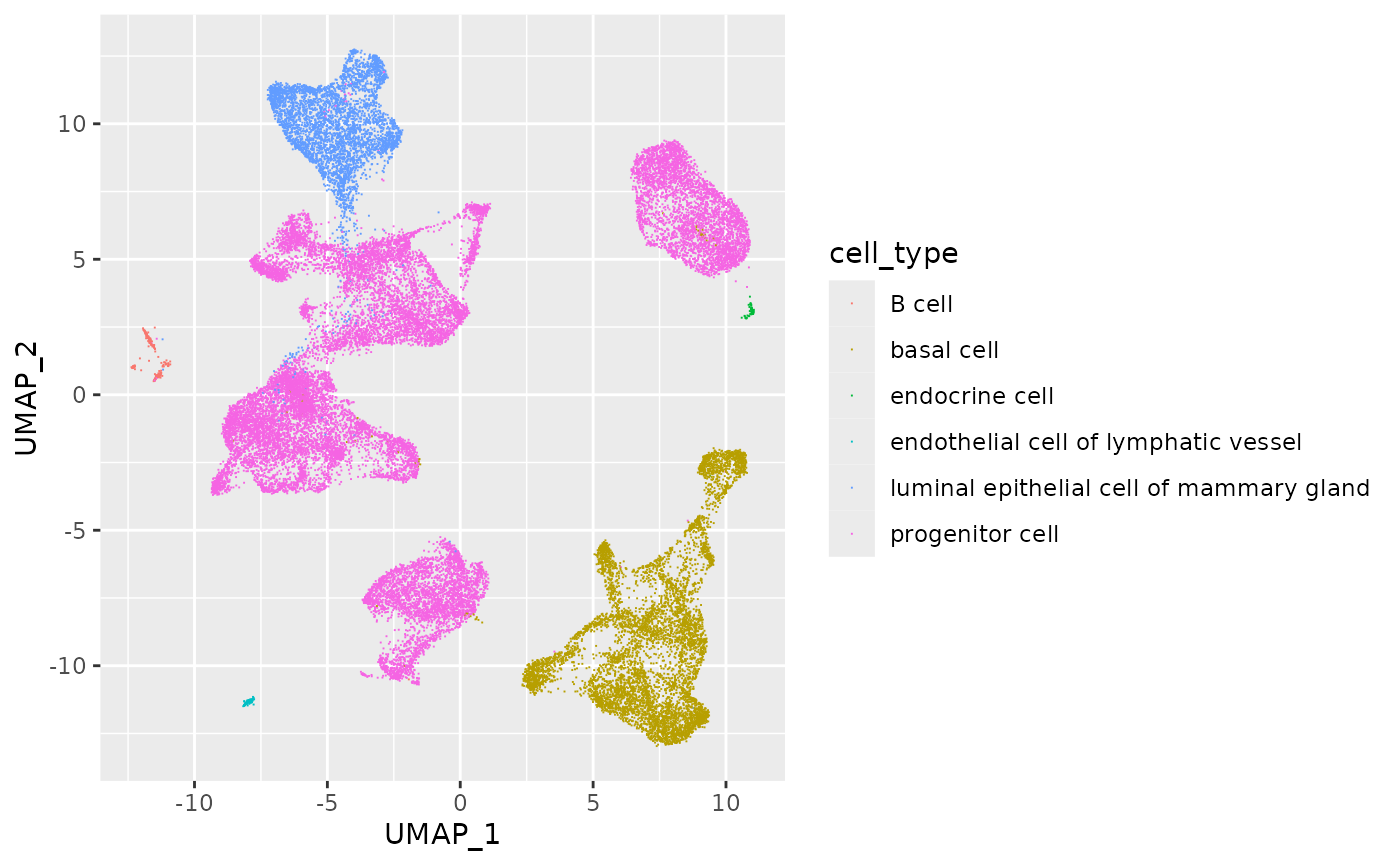
The plotly
package is a fantastic tool for making interactive plots, e.g., with
mouse-over ‘tool tips’ and ‘brushing’ selection. The
toWebGL() function makes display of plots with many points
very quick.
Survival analysis
These notes are from an online tutorial by Emily C. Zabor called Survival Analysis in R. In addition to dplyr and ggplot2, we use the specialized packages survival and ggsurvfit.
The main purpose of this case study is to introduce a statisitcal analysis that is common in oncology-related studies, and more nuanced than familiar statistics like a t-test or linear regression.
Data input, cleaning, & exploration
We use a data set that is built-in to R, available from the
survival
package. it’s a base R data.frame, but we’ll
coerce it immediately into a dplyr tibble.
lung <-
lung |>
as_tibble()
lung
## # A tibble: 228 × 10
## inst time status age sex ph.ecog ph.karno pat.karno meal.cal wt.loss
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 3 306 2 74 1 1 90 100 1175 NA
## 2 3 455 2 68 1 0 90 90 1225 15
## 3 3 1010 1 56 1 0 90 90 NA 15
## 4 5 210 2 57 1 1 90 60 1150 11
## 5 1 883 2 60 1 0 100 90 NA 0
## 6 12 1022 1 74 1 1 50 80 513 0
## 7 7 310 2 68 2 2 70 60 384 10
## 8 11 361 2 71 2 2 60 80 538 1
## 9 1 218 2 53 1 1 70 80 825 16
## 10 7 166 2 61 1 2 70 70 271 34
## # ℹ 218 more rowsThe meaning of each column is documented on the help page
?lung. We make two further changes. As noted on the help
page, status is coded as ‘1’ or ‘2’ (censored or dead; a
censored sample is an individual who is still alive at the end of the
study), but the usual convention is to code these as ‘0’ or ‘1’.
sex is coded as 1 or 2, but it seems much more sensible to
use a factor with levels ‘Female’ or ‘Male’)
lung <-
lung |>
mutate(
status = factor(
ifelse(status == 1, "Censored", "Death"),
levels = c("Censored", "Death")
),
sex = factor(
ifelse(sex == 1, "Male", "Female"),
levels = c("Female", "Male")
)
)The study contains fewer Female than Male samples; there is an
indication that mortality (status ‘1’) is higher in Male samples.
Deceased individuals are slightly older than censored individuals; not
surprisingly, the time (either until the end of the study
or death) is longer for censored individuals.
lung |>
count(sex, status) |>
tidyr::pivot_wider(names_from = "sex", values_from = "n")
## # A tibble: 2 × 3
## status Female Male
## <fct> <int> <int>
## 1 Censored 37 26
## 2 Death 53 112
lung |>
group_by(status) |>
summarize(n = n(), av_time = mean(time), av_age = mean(age))
## # A tibble: 2 × 4
## status n av_time av_age
## <fct> <int> <dbl> <dbl>
## 1 Censored 63 363. 60.3
## 2 Death 165 283 63.3Statistical analysis
The survival package
introduces a function Surv() that combines information
about time and status. Here are the first 6 rows of lung,
and the first 6 values returned by Surv()
lung |>
head()
## # A tibble: 6 × 10
## inst time status age sex ph.ecog ph.karno pat.karno meal.cal wt.loss
## <dbl> <dbl> <fct> <dbl> <fct> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 3 306 Death 74 Male 1 90 100 1175 NA
## 2 3 455 Death 68 Male 0 90 90 1225 15
## 3 3 1010 Censored 56 Male 0 90 90 NA 15
## 4 5 210 Death 57 Male 1 90 60 1150 11
## 5 1 883 Death 60 Male 0 100 90 NA 0
## 6 12 1022 Censored 74 Male 1 50 80 513 0
lung |>
with(Surv(time, status == "Death")) |>
head()
## [1] 306 455 1010+ 210 883 1022+Note the + associated with the third and sixth elements,
corresponding to status ‘0’ (censored) in
lung.
Use survfit() to fit a survival model to the data. The
model asks ‘what is the probability of survival to X number of days,
given the time and status observations?’. The
formula below, with ~ 1, indicates that we will not include
any co-variates, like the sex of samples.
fit <- survfit(Surv(time, status == "Death") ~ 1, lung)
fit
## Call: survfit(formula = Surv(time, status == "Death") ~ 1, data = lung)
##
## n events median 0.95LCL 0.95UCL
## [1,] 228 165 310 285 363A basic plot illustrates how probability of survival declines with time; dashed lines represent confidence intervals. A little more is covered below.
plot(fit)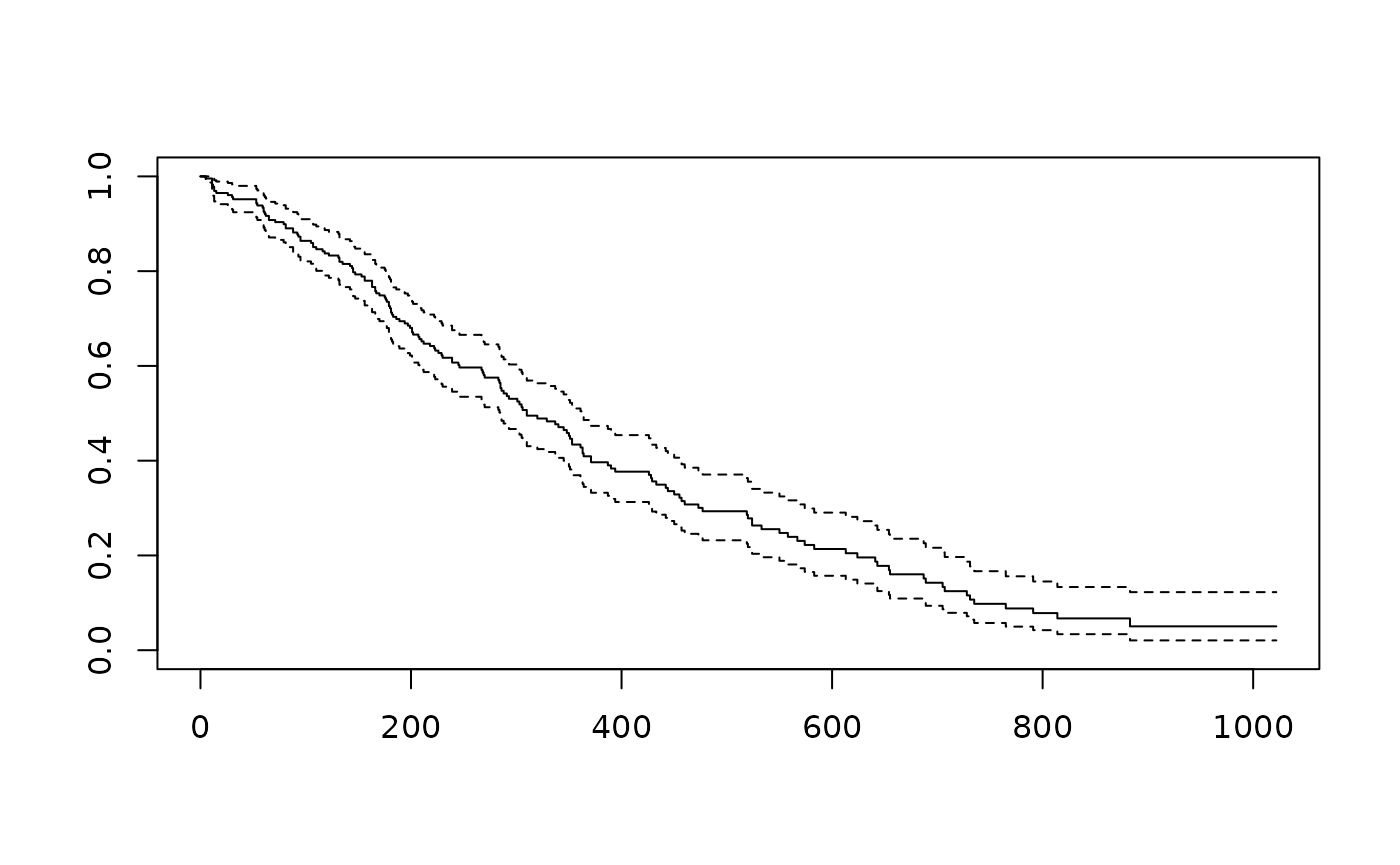
The fit object can be used to calculate various
statistics, e.g., the probability of surviving 180, 360, 540, or 720
days
days <- c(180, 360, 540, 720)
summary(fit, times = days)
## Call: survfit(formula = Surv(time, status == "Death") ~ 1, data = lung)
##
## time n.risk n.event survival std.err lower 95% CI upper 95% CI
## 180 160 63 0.722 0.0298 0.6655 0.783
## 360 70 54 0.434 0.0358 0.3693 0.510
## 540 33 26 0.255 0.0344 0.1962 0.333
## 720 14 15 0.125 0.0290 0.0789 0.197A more complicated model might ask about survival for each sex.
Construct this model by adding sex (a covariate) to the
right-hand side of the formula; subsequent steps are the same, but now
information is available for each sex.
fit_sex <- survfit(Surv(time, status == "Death") ~ sex, lung)
fit_sex
## Call: survfit(formula = Surv(time, status == "Death") ~ sex, data = lung)
##
## n events median 0.95LCL 0.95UCL
## sex=Female 90 53 426 348 550
## sex=Male 138 112 270 212 310
summary(fit_sex, times = days)
## Call: survfit(formula = Surv(time, status == "Death") ~ sex, data = lung)
##
## sex=Female
## time n.risk n.event survival std.err lower 95% CI upper 95% CI
## 180 71 14 0.842 0.0387 0.770 0.922
## 360 33 20 0.560 0.0586 0.457 0.688
## 540 16 10 0.368 0.0630 0.263 0.515
## 720 7 5 0.218 0.0641 0.123 0.388
##
## sex=Male
## time n.risk n.event survival std.err lower 95% CI upper 95% CI
## 180 89 49 0.6445 0.0408 0.569 0.730
## 360 37 34 0.3553 0.0437 0.279 0.452
## 540 17 16 0.1897 0.0385 0.127 0.282
## 720 7 10 0.0781 0.0276 0.039 0.156survdiff() is one way to test for the effect of
sex on survival.
survdiff(Surv(time, status == "Death") ~ sex, lung)
## Call:
## survdiff(formula = Surv(time, status == "Death") ~ sex, data = lung)
##
## N Observed Expected (O-E)^2/E (O-E)^2/V
## sex=Female 90 53 73.4 5.68 10.3
## sex=Male 138 112 91.6 4.55 10.3
##
## Chisq= 10.3 on 1 degrees of freedom, p= 0.001See also the Cox proportional hazards model.
Visualization
Functions in the ggsurvfit package provide ggplot2-style plotting functionality.
Here we plot the survivorship of Male and Female samples in the ‘lung’ data set, including confidence intervals.
survfit(Surv(time, status == "Death") ~ sex, lung) |>
ggsurvfit() +
add_confidence_interval()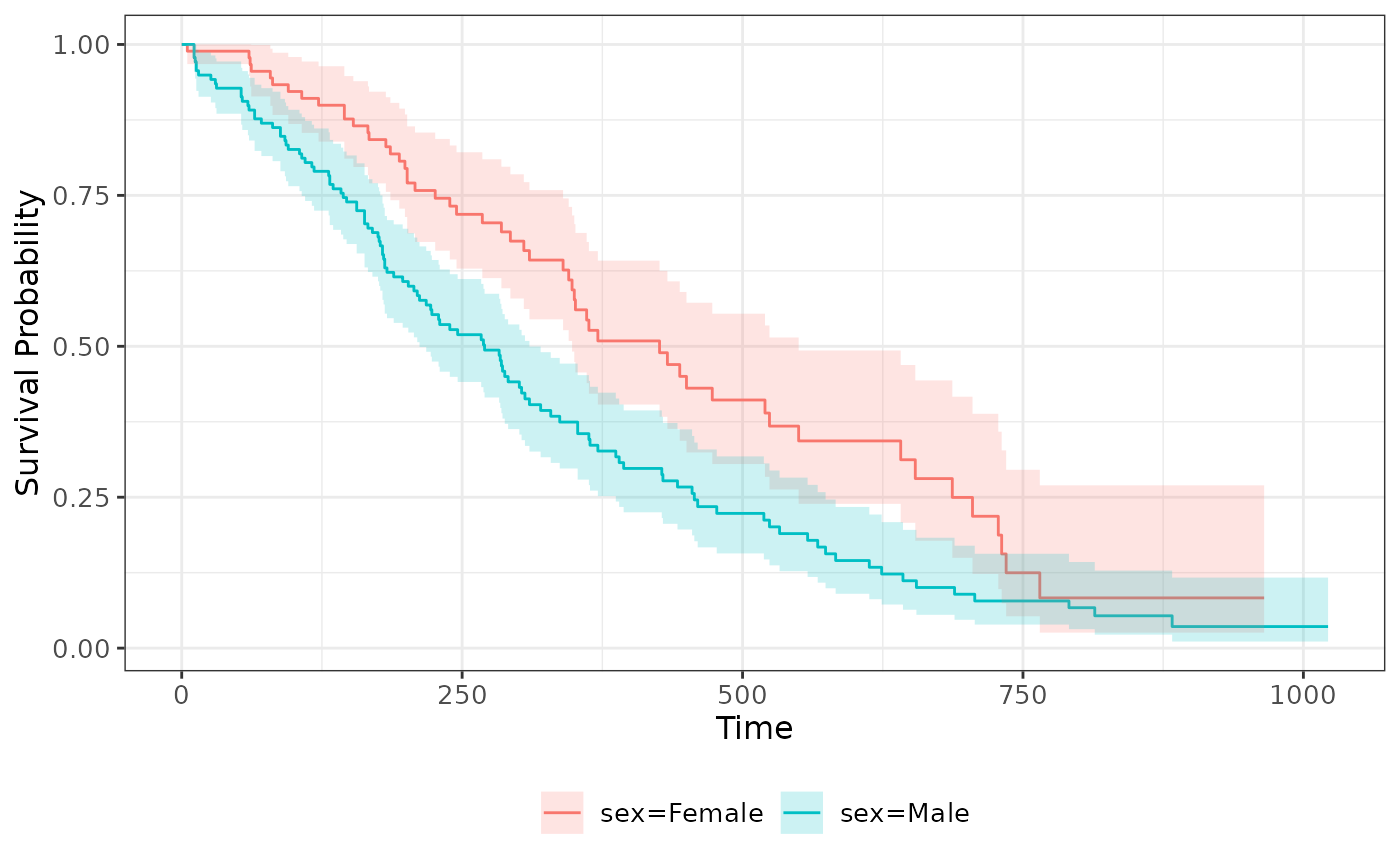
Next steps
The survival package contains a number of vignettes
vignette(package = "survival")
## Vignettes in package 'survival':
##
## adjcurve Adjusted Survival Curves (source, pdf)
## approximate Approximating a Cox Model (source, pdf)
## redistribute Brier scores and the redistribute-to-the-right
## algorithm (source, pdf)
## concordance Concordance (source, pdf)
## discrim Discrimination and Calibration (source, pdf)
## matrix Matrix exponentials (source, pdf)
## compete Multi-state models and competing risks (source,
## pdf)
## multi Multi-state survival curves (source, pdf)
## other Other vignettes (source, pdf)
## population Population contrasts (source, pdf)
## tiedtimes Roundoff error and tied times (source, pdf)
## splines Splines, plots, and interactions (source, pdf)
## survival The survival package (source, pdf)
## timedep Using Time Dependent Covariates (source, pdf)
## validate Validation (source, pdf)Start with the ‘survival’ vignette
vignette(package = "survival", "survival") if this is an
area of particular interest to you.
Session information
For reproducibility, I record the software versions used to create this document
sessionInfo()
## R version 4.3.3 (2024-02-29)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggsurvfit_1.0.0 survival_3.5-8 plotly_4.10.4 ggplot2_3.5.0
## [5] dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.4 xfun_0.42 bslib_0.6.1 htmlwidgets_1.6.4
## [5] lattice_0.22-5 tzdb_0.4.0 vctrs_0.6.5 tools_4.3.3
## [9] crosstalk_1.2.1 generics_0.1.3 parallel_4.3.3 tibble_3.2.1
## [13] fansi_1.0.6 highr_0.10 pkgconfig_2.0.3 Matrix_1.6-5
## [17] data.table_1.15.2 desc_1.4.3 lifecycle_1.0.4 compiler_4.3.3
## [21] farver_2.1.1 stringr_1.5.1 textshaping_0.3.7 munsell_0.5.0
## [25] htmltools_0.5.7 sass_0.4.8 yaml_2.3.8 lazyeval_0.2.2
## [29] pillar_1.9.0 pkgdown_2.0.7 crayon_1.5.2 jquerylib_0.1.4
## [33] tidyr_1.3.1 ellipsis_0.3.2 cachem_1.0.8 tidyselect_1.2.0
## [37] digest_0.6.34 stringi_1.8.3 purrr_1.0.2 labeling_0.4.3
## [41] splines_4.3.3 fastmap_1.1.1 grid_4.3.3 colorspace_2.1-0
## [45] cli_3.6.2 magrittr_2.0.3 utf8_1.2.4 broom_1.0.5
## [49] readr_2.1.5 withr_3.0.0 backports_1.4.1 scales_1.3.0
## [53] bit64_4.0.5 rmarkdown_2.25 httr_1.4.7 bit_4.0.5
## [57] ragg_1.2.7 hms_1.1.3 memoise_2.0.1 evaluate_0.23
## [61] knitr_1.45 viridisLite_0.4.2 rlang_1.1.3 glue_1.7.0
## [65] vroom_1.6.5 jsonlite_1.8.8 R6_2.5.1 systemfonts_1.0.5
## [69] fs_1.6.3