2. Accessing CellxGene Data
Martin Morgan1, Kayla Interdonato2, Yubo Cheng3.
Source:vignettes/B_CXG.Rmd
B_CXG.RmdOverview
R / Bioconductor packages used
The focus is on cellxgenedp package. This package emphasize dplyr and ‘tidy’ approaches to working with data.frames. Files downloaded from the HCA or CellXGene web sites can be imported into R / Bioconductor as SingleCellExperiment objects through the LoomExperiment and zellkonverter packages.
Workshop
Exploring the CellxGene web sites
The CellxGene Data Portal
Interactive visualization of CellxGene data sets
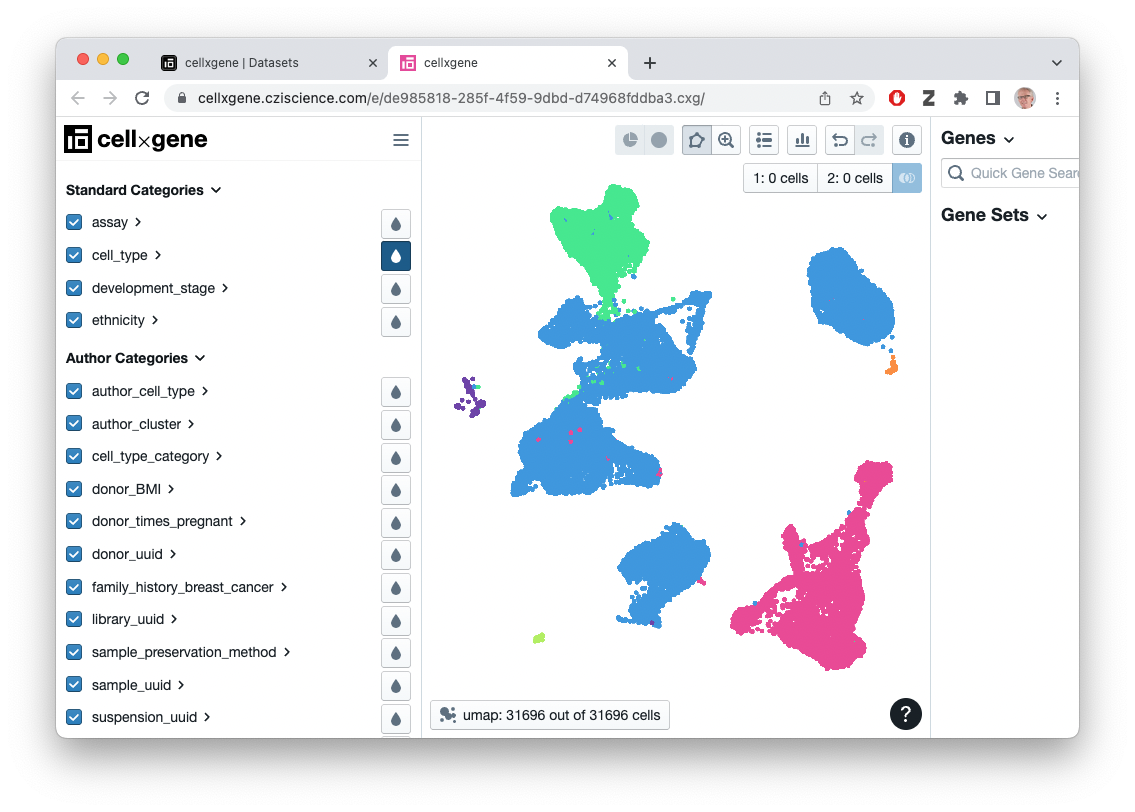
- Easy visualization is pretty cool!
Some observations
- Why is visualization fast? Pre-computed dimensionality reduction.
- What workflow was used in processing data? It’s up to the contributor, and not documented.
Working with CellxGene data
Load the packages we’ll use
suppressPackageStartupMessages({
library(cellxgenedp)
library(dplyr)
library(zellkonverter)
library(SingleCellExperiment)
library(ggplot2)
})Connect to the database
db = db()Discover datasets with 10x assays of African American females
african_american_female <-
datasets(db) |>
filter(
facets_filter(assay, "ontology_term_id", "EFO:0009922"),
facets_filter(ethnicity, "label", "African American"),
facets_filter(sex, "label", "female")
)
african_american_female
#> # A tibble: 21 × 27
#> dataset_id colle…¹ assay cell_…² cell_…³ datas…⁴ devel…⁵ disease ethni…⁶
#> <chr> <chr> <list> <int> <list> <chr> <list> <list> <list>
#> 1 de985818-285f… c9706a… <list> 31696 <list> https:… <list> <list> <list>
#> 2 f72958f5-7f42… 2f75d2… <list> 982538 <list> https:… <list> <list> <list>
#> 3 07854d9c-5375… bcb614… <list> 172847 <list> https:… <list> <list> <list>
#> 4 0b75c598-0893… bcb614… <list> 304652 <list> https:… <list> <list> <list>
#> 5 32b9bdce-2481… bcb614… <list> 107344 <list> https:… <list> <list> <list>
#> 6 59b69042-47c2… b9fc3d… <list> 49139 <list> https:… <list> <list> <list>
#> 7 e763ed0d-0e5a… b9fc3d… <list> 7274 <list> https:… <list> <list> <list>
#> 8 db0752b9-f20e… b9fc3d… <list> 55348 <list> https:… <list> <list> <list>
#> 9 d9b4bc69-ed90… b9fc3d… <list> 20000 <list> https:… <list> <list> <list>
#> 10 bc2a7b3d-f04e… b9fc3d… <list> 109995 <list> https:… <list> <list> <list>
#> # … with 11 more rows, 18 more variables: is_primary_data <chr>,
#> # is_valid <lgl>, linked_genesets <lgl>, mean_genes_per_cell <dbl>,
#> # name <chr>, organism <list>, processing_status <list>, published <lgl>,
#> # revision <int>, schema_version <chr>, sex <list>, tissue <list>,
#> # tombstone <lgl>, x_normalization <chr>, created_at <date>,
#> # published_at <date>, revised_at <date>, updated_at <date>, and abbreviated
#> # variable names ¹collection_id, ²cell_count, ³cell_type, …
#> # ℹ Use `print(n = ...)` to see more rows, and `colnames()` to see all variable namesFind H5AD files for these datasets; visualize and download one for illustration
selected_files <-
left_join(
african_american_female |> select(dataset_id),
files(db),
by = "dataset_id"
)
selected_files
#> # A tibble: 63 × 8
#> dataset_id file_id filen…¹ filet…² s3_uri user_…³ created_at updated_at
#> <chr> <chr> <chr> <chr> <chr> <lgl> <date> <date>
#> 1 de985818-285f-4… d5c6de… local.… H5AD s3://… TRUE 2021-09-24 2021-09-24
#> 2 de985818-285f-4… a0bcae… local.… RDS s3://… TRUE 2021-09-24 2021-12-21
#> 3 de985818-285f-4… dff842… explor… CXG s3://… TRUE 2021-09-24 2021-09-24
#> 4 f72958f5-7f42-4… 2e7374… local.… RDS s3://… TRUE 2021-10-07 2021-12-21
#> 5 f72958f5-7f42-4… bbce34… explor… CXG s3://… TRUE 2021-10-07 2021-10-07
#> 6 f72958f5-7f42-4… 091323… local.… H5AD s3://… TRUE 2021-10-07 2021-10-07
#> 7 07854d9c-5375-4… f6f812… local.… H5AD s3://… TRUE 2022-02-15 2022-02-15
#> 8 07854d9c-5375-4… 7ae7df… explor… CXG s3://… TRUE 2022-02-15 2022-02-15
#> 9 07854d9c-5375-4… 3f995f… local.… RDS s3://… TRUE 2022-02-15 2022-02-15
#> 10 0b75c598-0893-4… 38eaca… local.… H5AD s3://… TRUE 2022-02-15 2022-02-15
#> # … with 53 more rows, and abbreviated variable names ¹filename, ²filetype,
#> # ³user_submitted
#> # ℹ Use `print(n = ...)` to see more rowsVisualize ‘CXG’ files in the CellxGene web browser
selected_files |>
filter(filetype == "CXG") |>
dplyr::slice(1) |>
datasets_visualize()Download ‘H5AD’ (anndata) files for local use
h5ad_file <-
selected_files |>
filter(filetype == "H5AD") |>
dplyr::slice(1) |>
files_download(dry.run = FALSE)Importing .h5ad data into R / Bioconductor
Read the H5AD file into R
h5ad <- readH5AD(h5ad_file, reader = "R", use_hdf5 = TRUE)
h5ad
#> class: SingleCellExperiment
#> dim: 33234 31696
#> metadata(5): X_normalization default_embedding layer_descriptions
#> schema_version title
#> assays(1): X
#> rownames: NULL
#> rowData names(4): feature_biotype feature_is_filtered feature_name
#> feature_reference
#> colnames(31696): CMGpool_AAACCCAAGGACAACC CMGpool_AAACCCACAATCTCTT ...
#> K109064_TTTGTTGGTTGCATCA K109064_TTTGTTGGTTGGACCC
#> colData names(34): assay assay_ontology_term_id ...
#> tissue_ontology_term_id tyrer_cuzick_lifetime_risk
#> reducedDimNames(2): X_tsne X_umap
#> mainExpName: NULL
#> altExpNames(0):Integration with R / Bioconductor work flows
Work with the data, e.g., simple visualization to illustrate the familiar R / Bioconductor tools ‘just work’.
umap = as_tibble(reducedDim(h5ad, "X_umap"))
#> Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if `.name_repair` is omitted as of tibble 2.0.0.
#> Using compatibility `.name_repair`.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.
ggplot(umap) +
aes(x = V1, y = V2, color = h5ad$cell_type) +
geom_point(pch = ".")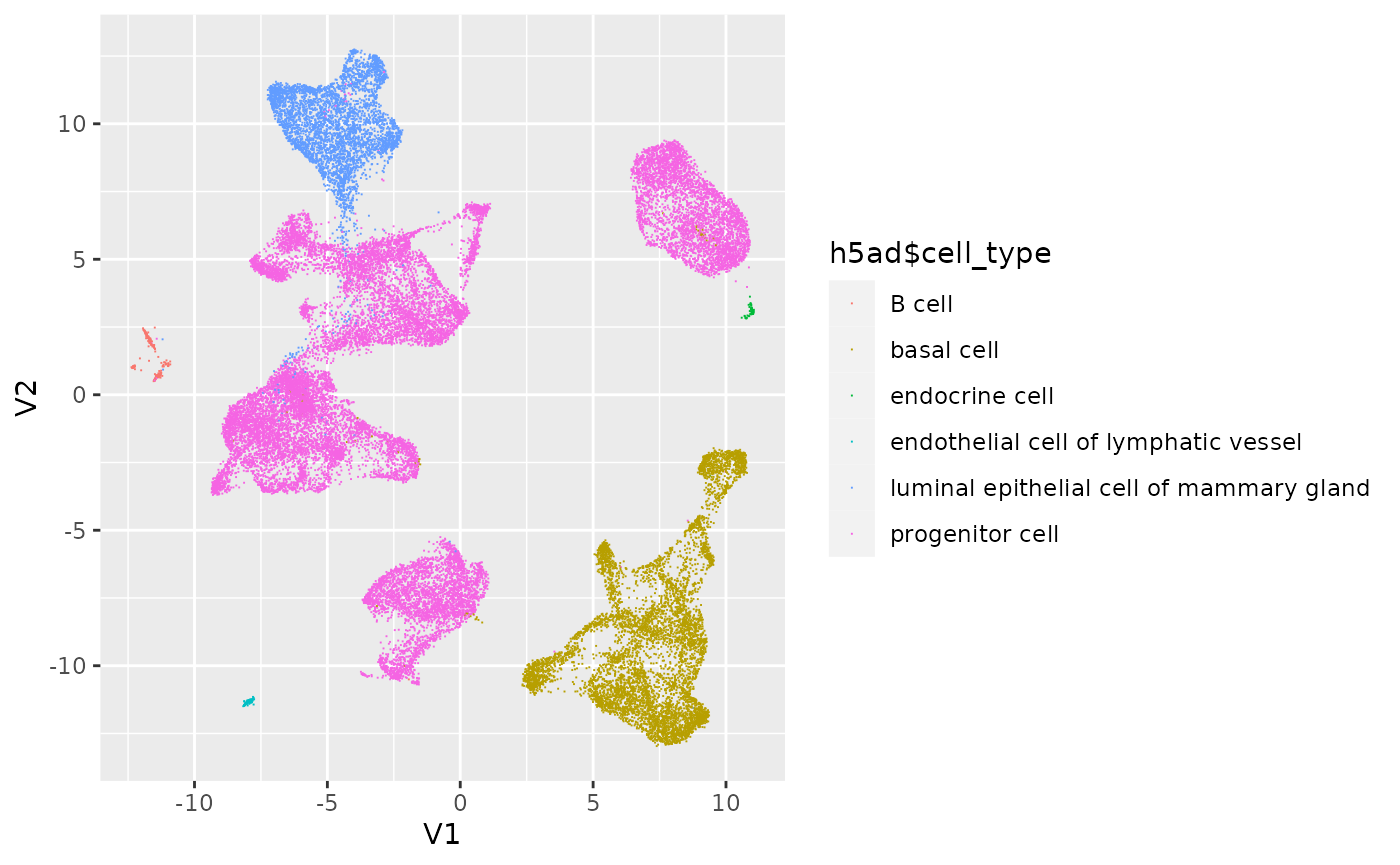
Acknowledgements
sessionInfo()
#> R version 4.2.0 (2022-04-22)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] ggplot2_3.3.6 SingleCellExperiment_1.19.0
#> [3] SummarizedExperiment_1.27.1 Biobase_2.57.1
#> [5] GenomicRanges_1.49.0 GenomeInfoDb_1.33.3
#> [7] IRanges_2.31.0 S4Vectors_0.35.1
#> [9] BiocGenerics_0.43.1 MatrixGenerics_1.9.1
#> [11] matrixStats_0.62.0 zellkonverter_1.7.3
#> [13] dplyr_1.0.9 cellxgenedp_1.1.2
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-7 fs_1.5.2 filelock_1.0.2
#> [4] httr_1.4.3 rprojroot_2.0.3 tools_4.2.0
#> [7] bslib_0.4.0 utf8_1.2.2 R6_2.5.1
#> [10] DT_0.23 HDF5Array_1.25.1 DBI_1.1.3
#> [13] colorspace_2.0-3 rhdf5filters_1.9.0 withr_2.5.0
#> [16] tidyselect_1.1.2 curl_4.3.2 compiler_4.2.0
#> [19] textshaping_0.3.6 cli_3.3.0 basilisk.utils_1.9.1
#> [22] desc_1.4.1 DelayedArray_0.23.0 labeling_0.4.2
#> [25] sass_0.4.2 scales_1.2.0 pkgdown_2.0.6
#> [28] systemfonts_1.0.4 stringr_1.4.0 digest_0.6.29
#> [31] rmarkdown_2.14 basilisk_1.9.2 XVector_0.37.0
#> [34] pkgconfig_2.0.3 htmltools_0.5.3 fastmap_1.1.0
#> [37] highr_0.9 htmlwidgets_1.5.4 rlang_1.0.4
#> [40] shiny_1.7.2 jquerylib_0.1.4 generics_0.1.3
#> [43] farver_2.1.0 jsonlite_1.8.0 RCurl_1.98-1.7
#> [46] magrittr_2.0.3 GenomeInfoDbData_1.2.8 Matrix_1.4-1
#> [49] Rcpp_1.0.9 munsell_0.5.0 Rhdf5lib_1.19.2
#> [52] fansi_1.0.3 reticulate_1.25 lifecycle_1.0.1
#> [55] stringi_1.7.8 yaml_2.3.5 zlibbioc_1.43.0
#> [58] rhdf5_2.41.1 grid_4.2.0 parallel_4.2.0
#> [61] promises_1.2.0.1 crayon_1.5.1 dir.expiry_1.5.0
#> [64] lattice_0.20-45 knitr_1.39 pillar_1.8.0
#> [67] glue_1.6.2 evaluate_0.15 vctrs_0.4.1
#> [70] png_0.1-7 httpuv_1.6.5 gtable_0.3.0
#> [73] purrr_0.3.4 assertthat_0.2.1 cachem_1.0.6
#> [76] xfun_0.31 mime_0.12 xtable_1.8-4
#> [79] later_1.3.0 ragg_1.2.2 tibble_3.1.7
#> [82] memoise_2.0.1 ellipsis_0.3.2